

Introduction
Materials and Methods
Reagents, inhibitors, and antibodies
Preparation of WSGL, WSGR, and WSGLR
Determination of ginsenoside contents in WSGL and WSGR
Cell culture
Griess assay for NO quantification
MTT assay for assessing WSGLR cytotoxicity in SIM-A9 cells
Reverse transcription polymerase chain reaction (RT-PCR) for iNOS mRNA in SIM-A9 cells
SDS-PAGE and immunoblotting
Measurement of SH-SY5Y cell viability under Aβ25-35 challenge
LDH release assay in SH-SY5Y cells
Measurement of intracellular ROS in SH-SY5Y cells
Statistical analysis
Results
Ginsenoside profiles of WSGL and WSGR
Comparative inhibition of LPS-induced NO production by WSGL, WSGR, and WSGLR in SIM-A9 microglia
Dose-dependent effects of WSGLR on LPS-induced NO production and iNOS expression in SIM-A9 microglia and its lack of cytotoxicity
WSGLR suppresses LPS-induced activation of MAPK and NF-κB signaling in SIM-A9 microglia
WSGLR induces nuclear Nrf2 accumulation and HO-1 expression in SIM-A9 microglia, and HO-1 inhibition attenuates WSGLR-mediated NO suppression
PI3K activity contributes to WSGLR-induced Nrf2 nuclear accumulation and HO-1 upregulation in SIM-A9 microglia
WSGLR protects SH-SY5Y cells against Aβ25-35-induced cytotoxicity
Discussion
Introduction
Neuroinflammation is now recognized as a core driver of neurodegenerative processes rather than a mere secondary consequence of neuronal injury. In Alzheimer’s disease (AD) and related disorders, chronic activation of microglia reshapes the brain milieu through sustained production of pro-inflammatory cytokines and reactive oxygen/nitrogen species, thereby amplifying synaptic dysfunction and accelerating neuronal loss (Lull and Block, 2010; Sarlus and Heneka, 2017). Importantly, amyloid-β (Aβ) pathology and microglial inflammatory signaling form self-reinforcing loops: Aβ can activate microglia through pattern-recognition and innate immune receptors, while microglia-derived inflammatory mediators can further exacerbate neuronal vulnerability and Aβ-associated toxicity (Lull and Block, 2010; Sarlus and Heneka, 2017).
Among microglia-derived inflammatory effectors, inducible nitric oxide synthase (iNOS) and its product nitric oxide (NO) have been widely used as mechanistically meaningful readouts of pro-inflammatory microglial activation. Excessive NO and related reactive nitrogen species contribute to oxidative/nitrosative stress, mitochondrial dysfunction, and downstream neuronal damage, particularly under conditions of prolonged glial activation (Brown and Bal-Price, 2003; Yuste et al., 2015). Toll-like receptor 4 (TLR4) stimulation by lipopolysaccharide (LPS) is a well-established experimental paradigm that induces NF-κB/MAPK-associated inflammatory cascades and robust iNOS upregulation in microglia, providing a reproducible platform to evaluate candidate anti-neuroinflammatory agents (Iizumi et al., 2016). In this context, SIM-A9 cells, a spontaneously immortalized murine microglial cell line, have been characterized as a practical microglia model that responds to LPS with activation of inflammatory signaling pathways and induction of iNOS, while retaining key microglial features (Nagamoto-Combs et al., 2014). Consistent with this, SIM-A9-based LPS activation systems have been used to quantify pharmacologic suppression of NO production as a functional index of microglial anti-inflammatory activity (Jamornwan et al., 2022).
In parallel, cell-intrinsic neuronal injury triggered by Aβ remains a canonical axis in AD-related neurotoxicity. While full-length Aβ peptides are central to AD pathology, the short fragment Aβ25-35 has been extensively used as a convenient, aggregation-prone neurotoxic peptide that induces apoptotic-like cell death and oxidative stress responses in neuronal cell models (Kim, 2014). Human neuroblastoma SH-SY5Y cells are frequently employed to examine neuroprotective mechanisms against Aβ-driven toxicity; exposure to Aβ25-35 has been reported to reduce SH-SY5Y viability in a dose- and time-dependent manner (Kim, 2014). Accordingly, a combined experimental strategy, LPS-stimulated microglia to model neuroinflammation and Aβ25-35-challenged neuronal cells to model amyloid-associated neurotoxicity, offers a rational framework to test whether a candidate material exerts both anti-neuroinflammatory and direct neuroprotective effects.
Panax ginseng and related ginseng resources have long attracted attention for multi-target neuroprotective potential, including anti-inflammatory, antioxidant, and pro-survival signaling effects relevant to neurodegeneration (Kim et al., 2018). Mechanistically, representative ginsenosides have been shown to suppress microglial inflammatory mediator production and to interfere with key signaling nodes such as MAPKs and NF-κB, supporting the concept that ginseng-derived phytochemicals can modulate neuroinflammatory activation states (Madhi et al., 2021). At the same time, the chemical composition of ginseng is highly tissue-dependent. Multiple analyses have demonstrated that ginsenoside abundance and profiles differ across plant parts, with leaves and root hairs often containing higher total ginsenosides than the main root and showing distinct patterns of specific ginsenosides (Kang and Kim, 2016). More broadly, stem-leaf tissues of Panax species have been proposed as a rich and sustainable source of ginsenosides, underscoring the scientific rationale for incorporating aerial tissues rather than restricting extraction solely to roots (Zhang et al., 2021). In addition to saponins, Panax polysaccharides have been reviewed as bioactive fractions with immunomodulatory actions, further motivating the evaluation of composite extracts that may capture complementary classes of constituents (Hu et al., 2021). Notably, for wild-simulated ginseng, comparative profiling of aerial (leaf/stem) versus underground (rhizome/radix) parts has also been reported, highlighting measurable differences in chemical composition between the parts and supporting the feasibility of designing mixed-part extracts with broader phytochemical coverage (Roh et al., 2023).
Despite these lines of evidence, studies evaluating a blended mixture prepared by separately extracting the aerial and underground parts of wild simulated ginseng and then combining the dried extracts at a 1:1 ratio (w/w), while simultaneously assessing microglial iNOS/NO suppression under LPS-mediated activation and direct neuroprotection against Aβ25-35-induced neuronal cell death, remain limited. Therefore, the present study investigated a wild simulated ginseng complex prepared by mixing extracts of the aerial and underground parts at a 1:1 ratio. Using LPS-stimulated SIM-A9 microglia, we assessed inhibition of iNOS expression and NO production as primary anti-neuroinflammatory endpoints. In SH-SY5Y neuronal cells challenged with Aβ25-35, we evaluated the ability of the extract to attenuate amyloid-fragment-induced cytotoxicity. Together, this dual-model approach was designed to clarify whether an aerial-underground mixed extract of wild simulated ginseng can provide convergent anti-neuroinflammatory and neuroprotective activities relevant to neurodegeneration-associated inflammatory and amyloidogenic insults.
Materials and Methods
Reagents, inhibitors, and antibodies
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; cat. no. 475989), LY294002 (cat. no. 440202), Griess reagent (cat. no. G4410), lipopolysaccharide (LPS; cat. no. L2630), and Aβ25-35 (cat. no. A4559) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Zinc(II) protoporphyrin IX (ZnPP; cat. no. ALX-430-049-M025) was purchased from Enzo Life Sciences (Long Island, NY, USA). Primary antibodies for immunoblotting were sourced as follows. Antibodies against ERK1/2 (cat. no. 9102), phospho-ERK1/2 (cat. no. 4377), p38 (cat. no. 9212), phospho-p38 (cat. no. 4511), JNK (cat. no. 9258), phospho-JNK (cat. no. 9251), p65 (cat. no. 8242), phospho-p65 (cat. no. 3033), PI3K (cat. no. 4257), phospho-PI3K (cat. no. 4228), HO-1 (cat. no. 70081), Nrf2 (cat. no. 12721), and β-actin (cat. no. 5125) were obtained from Cell Signaling Technology (Danvers, MA, USA). Antibodies against Bcl-2 (cat. no. sc-7382) and Bax (cat. no. sc-7480) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Horseradish peroxidase (HRP)-conjugated secondary antibodies such as anti-rabbit IgG (cat. no. 7074) and anti-mouse IgG (cat. no. 7076) were obtained from Cell Signaling Technology.
Preparation of WSGL, WSGR, and WSGLR
Wild-simulated ginseng was separated into aerial parts (stems and leaves) and underground parts (roots). Each plant portion was cleaned independently to remove adhering debris, freeze-dried, and pulverized into a fine powder. The powdered aerial material was extracted to obtain the aerial-part extract (WSGL), and the powdered underground material was extracted in parallel to obtain the underground-part extract (WSGR) using the same extraction solvent system and conditions to minimize process-derived compositional bias. Briefly, each powder was mixed with 50% ethanol at a fixed solid-to-solvent ratio (1:20), extracted at 20°C for 24 h with agitation, and then clarified by filtration. The filtrates were concentrated under reduced pressure and freeze-dried to yield solid extracts. The resulting WSGL and WSGR powders were stored in airtight containers protected from light and moisture at -80°C until use. To prepare the blended formulation (WSGLR), WSGL and WSGR were weighed based on dry extract mass and combined at a 1:1 (w/w) ratio. The mixture was homogenized by thorough vortexing and repeated gentle mixing until a visually uniform powder was obtained. For cell-based experiments, each extract (WSGL, WSGR) and the blend (WSGLR) were freshly prepared as a concentrated stock in DMSO. The final solvent content was kept constant across treatment groups and matched in vehicle controls.
Determination of ginsenoside contents in WSGL and WSGR
The ginsenoside contents of WSGL and WSGR were analyzed by the Seoul National University National Instrumentation Center for Environmental Management (NICEM, Seoul, Republic of Korea) as a fee-for-service measurement. Dried WSGL and WSGR powders were submitted to NICEM for targeted ginsenoside profiling and quantification according to the facility’s internal standard operating procedures (SOPs). The analytical results were provided by the service laboratory as compound identities and corresponding quantitative values, with non-detected compounds reported as ND.
Cell culture
Murine microglial SIM-A9 cells (cat. no. CRL-3265) and human neuroblastoma SH-SY5Y cells (cat. no. CRL-2266) were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were routinely propagated at 37°C in a humidified incubator with 5% CO2. Both cell lines were cultured in DMEM/F-12 medium containing 10% fetal bovine serum (FBS) and penicillin-streptomycin (final concentrations: 100 U/mL penicillin and 100 ㎍/mL streptomycin). Culture medium was replaced at regular intervals, and cells were subcultured at an appropriate confluence using standard aseptic procedures.
Griess assay for NO quantification
NO production was assessed by measuring nitrite accumulation in culture supernatants using the Griess reaction. SIM-A9 cells were seeded in 12-well plates and allowed to attach until they reached 80-90% confluence. Cells were then exposed to WSGL, WSGR, or WSGLR at the indicated concentrations (screening: 100 ㎍/mL, dose-response: 50, 100, and 200 ㎍/mL) for 2 h and subsequently challenged with LPS (final concentration: 1 ㎍/mL) for 18 h. Vehicle controls (DMSO) were prepared to match the final solvent content in all treatment groups. At the end of stimulation, culture supernatants were collected and mixed with Griess reagent at an equal volume ratio (1:1, v/v) in a new plate. After incubation at room temperature for 15 min protected from light, absorbance was read at 540 ㎚ using a microplate reader (SpectraMax M2, Molecular Devices, San Jose, CA, USA).
MTT assay for assessing WSGLR cytotoxicity in SIM-A9 cells
Cell viability following WSGLR exposure was evaluated using the MTT assay. SIM-A9 cells were seeded into 96-well plates and allowed to attach until they reached 80-90% confluence. Cells were then treated with WSGLR at the indicated concentrations (50, 100, and 200 ㎍/mL) for 24 h. Vehicle controls (DMSO) were prepared to match the final solvent content in all treatment groups. After treatment, MTT solution (final concentration: 0.5 ㎎/mL) was added to each well, and the plates were incubated at 37°C for 4 h to allow formazan formation. The medium was carefully removed, and the resulting formazan crystals were dissolved in DMSO with gentle shaking until the solution became homogeneous. Absorbance was read at 570 ㎚ using a microplate reader (SpectraMax M2, Molecular Devices, San Jose, CA, USA).
Reverse transcription polymerase chain reaction (RT-PCR) for iNOS mRNA in SIM-A9 cells
LPS-induced transcriptional upregulation of iNOS in SIM-A9 cells and its modulation by WSGLR were examined by RT-PCR. SIM-A9 cells were seeded in 6-well plates and cultured to 80-90% confluence. Cells were pre-treated with WSGLR (100 and 200 ㎍/mL) for 2 h, followed by stimulation with LPS (final concentration: 1 ㎍/mL) for 18 h. Vehicle controls (DMSO) were included by matching the final solvent content across all groups. After treatment, total RNA was isolated using a silica membrane-based spin-column kit (RNeasy Mini Kit, Qiagen, Hilden, Germany) according to the manufacturer’s instructions. RNA quantity and purity were assessed by spectrophotometry (A260/A280), and equal amounts of total RNA (1 ㎍ per sample) were reverse-transcribed to cDNA using a commercial reverse transcription kit (Verso cDNA Kit, Thermo Fisher Scientific, Waltham, MA, USA). The resulting cDNA was amplified using a premixed PCR reagent (PCR Master Mix, Promega, Madison, WI, USA) with gene-specific primers for iNOS and the housekeeping gene GAPDH. The primer sequences were as follows: iNOS, forward 5′-GTTACCATGAGGCTGAAATCC-3′ and reverse 5′-CCTCTTGTCTTTGACCCAGTAC-3′; GAPDH, forward 5′-GGACCTCATGGCCTACATGG-3′ and reverse 5′-TAGGGCCTCTCTTGCTCAGT-3′. PCR products were resolved on 1 % agarose gels and visualized under UV illumination. Band intensities were quantified using gel densitometry software (UN-SCAN-IT Gel v5.1, Silk Scientific Inc., Vineyard, UT, USA), and iNOS signals were normalized to GAPDH.
SDS-PAGE and immunoblotting
After the designated treatments, whole-cell lysates were prepared by solubilizing cells in ice-cold RIPA lysis buffer (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with protease and phosphatase inhibitor cocktails. Lysates were clarified by centrifugation at 15,000 × g for 30 min at 4°C, and protein concentrations were determined using a bicinchoninic acid (BCA) assay (Pierce™ BCA Protein Assay Kit, Thermo Fisher Scientific, Waltham, MA, USA). Equal amounts of protein (30 ㎍/well) were mixed with SDS sample buffer, denatured at 95°C for 10 min, and separated by SDS-polyacrylamide gel electrophoresis on 10% gels. Proteins were transferred to nitrocellulose membranes (Thermo Fisher Scientific, Waltham, MA, USA) using a wet transfer system under 100 V/300 A for 2 h. Membranes were blocked in 5% (w/v) non-fat dry milk prepared in TBST for 1 h at room temperature and then incubated with primary antibodies (1:1,000) overnight at 4°C. After washing with TBST, membranes were incubated with HRP-conjugated secondary antibodies (1:1,000) for 1 h at room temperature. Immunoreactive bands were visualized using ECL Prime Western Blotting Detection Reagents (Amersham Biosciences Corp., Piscataway, NJ, USA) and captured with a digital blot imaging system (C-DiGit Blot Scanner, LI-COR Biosciences, Lincoln, NE, USA). Band intensities were quantified by densitometry using UN-SCAN-IT Gel v5.1 (Silk Scientific Inc., Vineyard, UT, USA). For normalization, β-actin was used as a loading control.
Measurement of SH-SY5Y cell viability under Aβ25-35 challenge
Cell viability in SH-SY5Y cells exposed to Aβ25-35 was assessed using an MTT assay. SH-SY5Y cells were seeded into 12-well plates and allowed to attach until they reached 80-90% confluence. Cells were then pre-treated with WSGLR at the indicated concentrations (50, 100, and 200 ㎍/mL) for 2 h, followed by exposure to Aβ25-35 (final concentration: 30 μM) for 24 h under standard incubation conditions (37°C, 5% CO2). Vehicle controls (DMSO) were included by matching the final solvent content in all wells. At the end of treatment, the culture medium was removed and replaced with fresh medium containing MTT to a final concentration of 0.5 ㎎/mL. Plates were incubated for 4 h to allow intracellular reduction of MTT and formation of formazan. The MTT-containing medium was then aspirated, and the formazan crystals were solubilized in DMSO with gentle shaking until fully dissolved. Absorbance was recorded at 570 ㎚ using a microplate reader (SpectraMax M2, Molecular Devices, San Jose, CA, USA).
LDH release assay in SH-SY5Y cells
Membrane damage-associated cytotoxicity was evaluated by quantifying lactate dehydrogenase (LDH) activity released into the culture supernatant. SH-SY5Y cells were plated in 12-well] plates and allowed to reach 80-90% confluence. Cells were pre-treated with WSGLR (50, 100, and 200 ㎍/mL) and then challenged with Aβ25-35 (final concentration: 30 μM) for 24 h under standard culture conditions (37°C, 5% CO2). Vehicle controls (DMSO) were included by matching the final solvent content across all wells. Following treatment, culture supernatants were collected, and LDH release was determined using the CyQUANT™ LDH Cytotoxicity Assay Kit (Invitrogen Co., Carlsbad, CA, USA) in accordance with the manufacturer’s instructions.
Measurement of intracellular ROS in SH-SY5Y cells
Intracellular reactive oxygen species (ROS) generation in SH-SY5Y cells was assessed using a fluorometric ROS detection kit. SH-SY5Y cells were seeded in 12-well plates and cultured until 80-90% confluence. Cells were pre-exposed to WSGLR at the indicated concentrations (100 and 200 ㎍/mL), followed by Aβ25-35 challenge (final concentration: 30 μM) for 24 h under standard incubation conditions (37°C, 5% CO2). Vehicle controls (DMSO) were included with an identical final solvent content. After treatment, intracellular ROS levels were quantified using the Intracellular ROS Assay Kit (CELL BIOLABS, INC., San Diego, CA, USA) according to the manufacturer’s instructions.
Statistical analysis
All experiments were repeated at least three times. Statistical analysis was performed using GraphPad Prism version 5.0 (GraphPad Software, Inc.), and data are presented as the mean ± standard deviation. Data was analyzed using one-way analysis of variance followed by Bonferroni’s post hoc test. P<0.05 was considered to indicate a statistically significant difference.
Results
Ginsenoside profiles of WSGL and WSGR
Targeted ginsenoside profiling revealed clear quantitative and qualitative differences between WSGL and WSGR (Table 1). Several ginsenosides were detected in only one of the two samples. Specifically, F3, F5, and Rd2 were detected in WSGL but not in WSGR (ND), whereas Ra1, Ra2, and Ra3 were detected in WSGR but not in WSGL (ND). Among the quantified ginsenosides, Re was the most abundant component in both WSGL (19,436.78 ± 634.89 ㎍/g extract) and WSGR (6,292.22 ± 397.45 ㎍/g extract). WSGL contained high levels of multiple F-series and protopanaxadiol-type ginsenosides, including F1 (4,040.03 ± 12.705 ㎍/g extract), F2 (126.57 ± 10.65 ㎍/g extract), F3 (621.59 ± 60.63 ㎍/g extract), F5 (4,741.49 ± 649.06 ㎍/g extract), and Rd (9,677.44 ± 271.57 ㎍/g extract). In contrast, WSGR was characterized by substantial amounts of Ra2 (12,341.69 ± 946.94 ㎍/g extract) and Rb1 (8,576.68 ± 727.44 ㎍/g extract), as well as detectable levels of Ra1 (1,201.28 ± 212.77 ㎍/g extract) and Ra3 (668.96 ± 293.49 ㎍/g extract). Marked differences in abundance were observed for several ginsenosides. For example, Rb1 was higher in WSGR than in WSGL (8,576.68 ± 727.44 ㎍/g extract vs 922.13 ± 133.12 ㎍/g extract), whereas Rd was higher in WSGL than in WSGR (9,677.44 ± 271.57 ㎍/g extract vs 344.03 ± 16.07 ㎍/g extract).
Table 1.
Targeted ginsenoside profiles of WSGL and WSGR.
Comparative inhibition of LPS-induced NO production by WSGL, WSGR, and WSGLR in SIM-A9 microglia
Because the targeted ginsenoside analysis demonstrated clear part-specific profiles-i.e., the presence of several ginsenosides in only one of the two extracts (Table 1), the anti-neuroinflammatory activity of WSGL, WSGR, and their 1:1 (w/w) blended mixture (WSGLR) was compared by quantifying LPS-induced NO production in SIM-A9 microglial cells (Fig. 1). We additionally evaluated WSGLR to determine whether combining the two extracts provides a functional advantage in suppressing LPS-driven inflammatory output in microglia. At a single test concentration of 100 μg/mL, LPS treatment (DMSO vehicle) increased NO production to 3.01-fold relative to the untreated control (CON). In the presence of LPS, treatment with WSGL reduced NO production to 2.29-fold of CON, whereas WSGR reduced NO production to 1.55-fold of CON. Notably, the blended mixture WSGLR further reduced NO production to 1.15-fold of CON (Fig. 1). When expressed relative to the LPS-only group, NO levels were decreased by approximately 24% (WSGL), 49% (WSGR), and 62% (WSGLR) under the same conditions. Based on this comparative screening result, WSGLR was selected for subsequent experiments in this study.
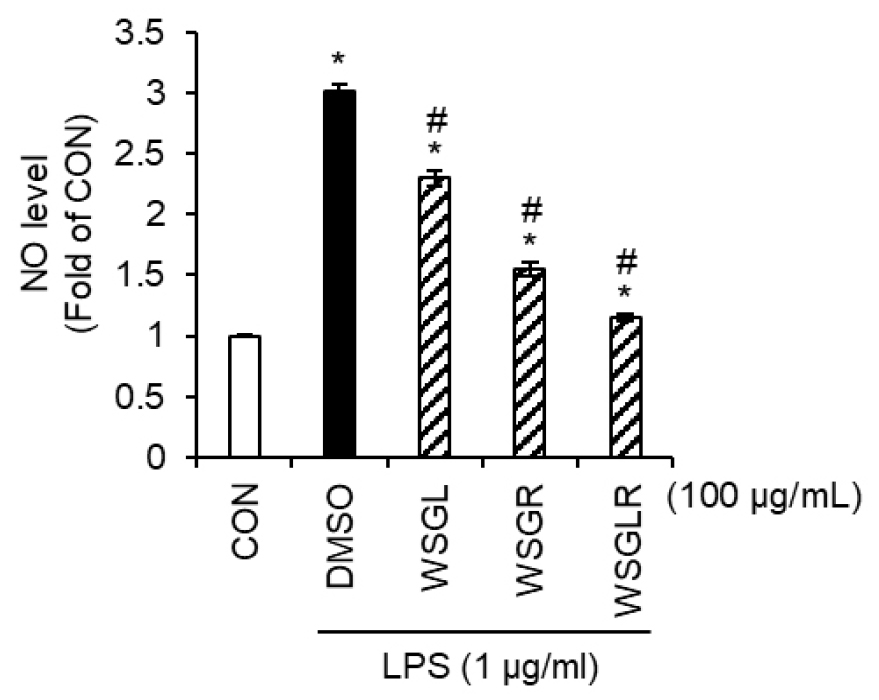
Fig. 1.
Comparative inhibition of LPS-induced NO production by WSGL, WSGR, and WSGLR in SIM-A9 microglia. SIM-A9 cells were treated with the wild-simulated ginseng aerial-part extract (WSGL), underground-part extract (WSGR), or their 1:1 (w/w) blended mixture (WSGLR) at 100 ㎍/mL in the presence of LPS. NO production was quantified by measuring nitrite accumulation in culture supernatants using the Griess reaction. Data are presented as fold change relative to the untreated control (CON), with the LPS-only group receiving vehicle (DMSO). Values are shown as mean ± SD from three independent experiments. *p < 0.05 vs. CON, #P < 0.05 vs. LPS-only (DMSO).
Dose-dependent effects of WSGLR on LPS-induced NO production and iNOS expression in SIM-A9 microglia and its lack of cytotoxicity
Based on the comparative screening showing the strongest suppression of LPS-induced NO production by WSGLR (Fig. 1), WSGLR was carried forward for concentration-response evaluation in SIM-A9 microglial cells (Fig. 2). In the LPS-stimulated condition, the vehicle control (DMSO group) increased NO production to 3.01-fold relative to the untreated control (CON) (Fig. 2A). In the presence of LPS, WSGLR reduced NO production in a concentration-dependent manner: NO levels were 2.47-fold, 1.45-fold, and 1.18-fold of CON at 50, 100, and 200 ㎍/mL, respectively (Fig. 2A). Consistent with the NO results, LPS robustly induced iNOS expression to 5.56-fold of CON in the DMSO group (Fig. 2B). Co-treatment with WSGLR suppressed iNOS expression at the tested concentrations, decreasing iNOS levels to 2.98-fold of CON at 100 ㎍/mL and further to 1.27-fold of CON at 200 μg/mL (Fig. 2B). In parallel, WSGLR did not reduce SIM-A9 cell viability at 50-200 ㎍/mL, indicating no detectable cytotoxicity under the experimental conditions (Fig. 2C).
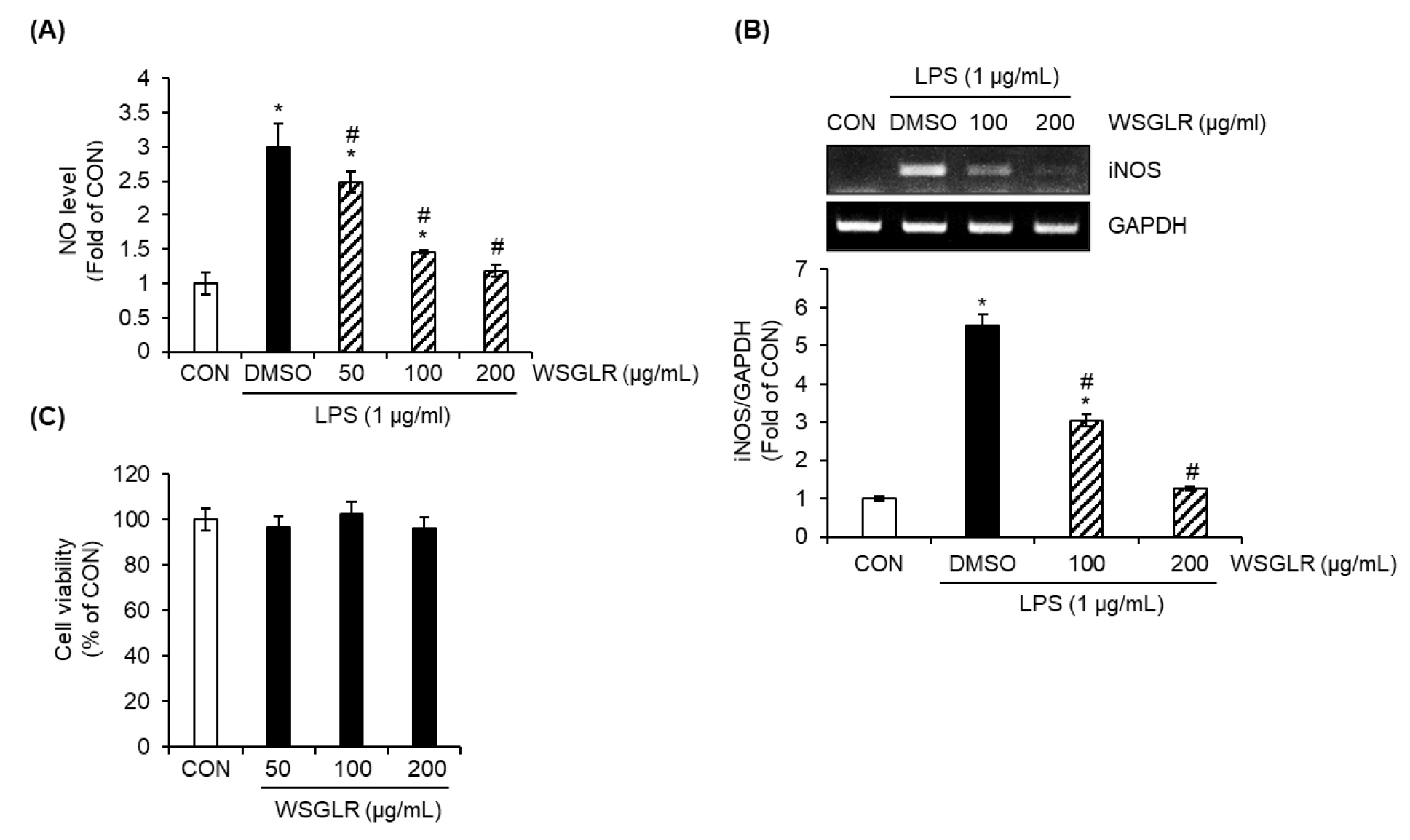
Fig. 2.
Dose-dependent suppression of LPS-induced NO production and iNOS expression by WSGLR in SIM-A9 microglia without detectable cytotoxicity. SIM-A9 cells were treated with WSGLR (50, 100, or 200 ㎍/mL) in the presence of LPS, with vehicle (DMSO) included as the LPS-only control and untreated cells serving as the control (CON). (A) NO production was determined by measuring nitrite levels in culture supernatants using the Griess assay and is presented as fold change relative to CON. (B) iNOS protein expression was analyzed by RT-PCR, quantified by densitometry, and expressed as fold change relative to CON after normalization to GAPDH. (C) Cell viability following WSGLR treatment was assessed by the MTT assay and is expressed as a percentage of CON. Data are presented as mean ± SD from three independent experiments. *p < 0.05 vs. CON, #P < 0.05 vs. LPS-only (DMSO).
WSGLR suppresses LPS-induced activation of MAPK and NF-κB signaling in SIM-A9 microglia
Given that WSGLR reduced LPS-induced NO production and iNOS expression without detectable cytotoxicity in SIM-A9 cells (Fig. 2), we next examined whether WSGLR modulates upstream inflammatory signaling pathways implicated in LPS-driven microglial activation (Fig. 3). In the LPS-stimulated condition, the vehicle control (DMSO group) markedly increased the phosphorylation ratios of ERK1/2 (p-ERK1/2/ERK1/2), p38 (p-p38/p38), and JNK (p-JNK/JNK) relative to the untreated control (CON) (Fig. 3A), indicating robust activation of MAPK signaling. In the presence of LPS, WSGLR reduced p-p38/p38 and p-JNK/JNK compared with the LPS-only group, whereas p-ERK1/2/ERK1/2 was not noticeably altered by WSGLR under the same conditions (Fig. 3A). Consistent with these MAPK findings, LPS (DMSO group) also increased NF-κB p65 phosphorylation (p-p65/p65) compared with CON (Fig. 3B). Co-treatment with WSGLR attenuated the LPS-induced increase in p-p65/p65 relative to the LPS-only group (Fig. 3B).
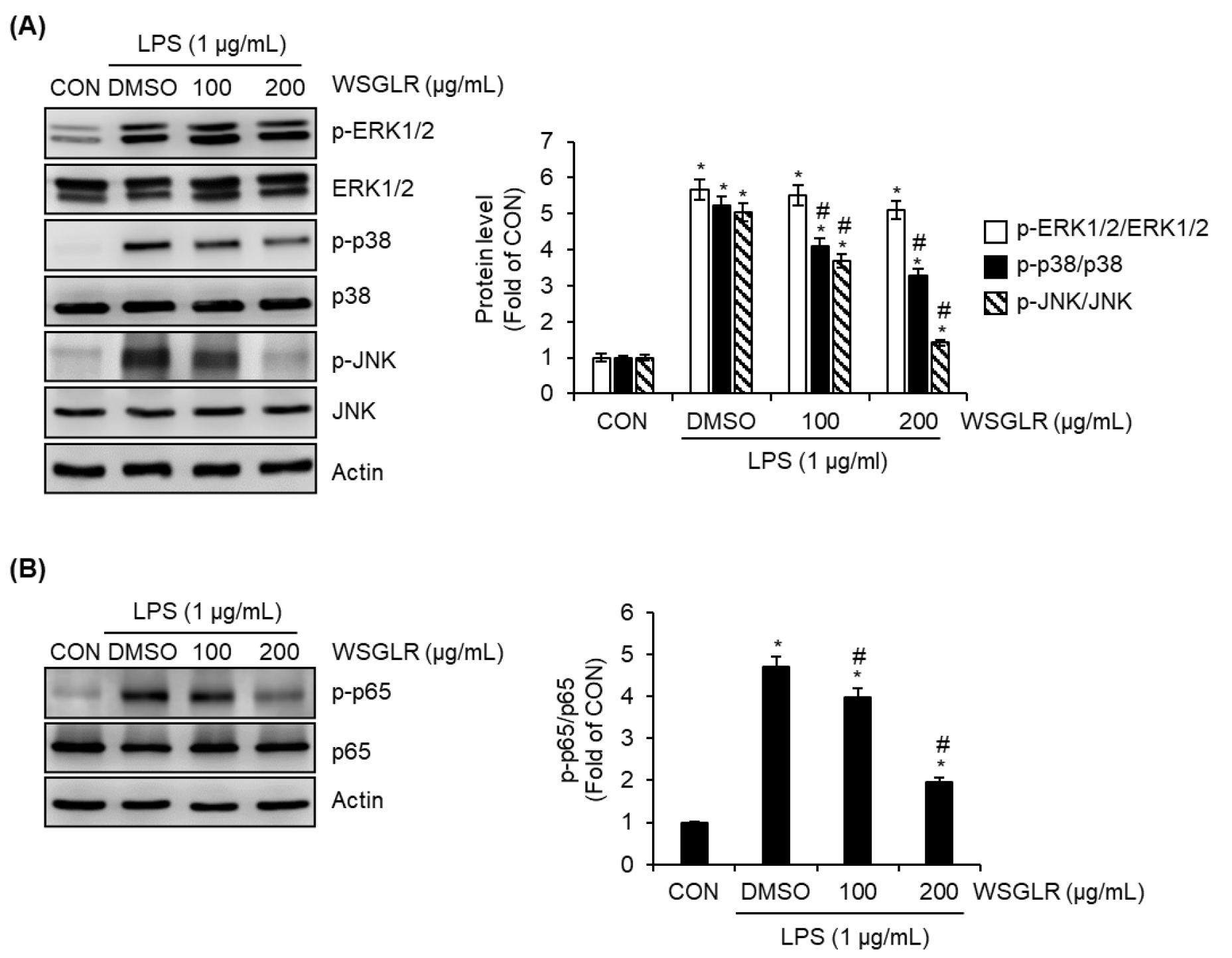
Fig. 3.
WSGLR attenuates LPS-induced activation of MAPK and NF-κB signaling in SIM-A9 microglia. SIM-A9 cells were treated with WSGLR (100 and 200 ㎍/mL) in the presence of LPS; vehicle (DMSO) served as the LPS-only control, and untreated cells were used as the control (CON). (A) Activation of MAPK signaling was assessed by immunoblotting for phosphorylated and total ERK1/2, p38, and JNK, and the phosphorylation ratios (p-ERK1/2/ERK1/2, p-p38/p38, and p-JNK/JNK) were quantified by densitometry. (B) NF-κB activation was evaluated by immunoblotting for phosphorylated and total p65, and the p-p65/p65 ratio was quantified. Data are presented as mean ± SD from three independent experiments. *p < 0.05 vs. CON, #P < 0.05 vs. LPS-only (DMSO).
WSGLR induces nuclear Nrf2 accumulation and HO-1 expression in SIM-A9 microglia, and HO-1 inhibition attenuates WSGLR-mediated NO suppression
Consistent with the observed suppression of LPS-driven inflammatory signaling (Fig. 3), we further examined whether WSGLR modulates the cytoprotective Nrf2/HO-1 pathway in SIM-A9 microglia (Fig. 4). Time-course analysis showed that treatment with WSGLR (200 ㎍/mL) produced a delayed but clear induction of HO-1 protein. HO-1 levels were not markedly altered at 1-3 h, whereas HO-1 began to increase significantly from 6 h onward and remained elevated at later time points up to 24 h (Fig. 4A). In contrast to the delayed HO-1 induction, nuclear Nrf2 levels increased rapidly following WSGLR exposure. When SIM-A9 cells were treated with WSGLR (200 ㎍/mL) for 1, 3, and 6 h, nuclear Nrf2 was noticeably elevated as early as 1 h, indicating early activation of Nrf2 signaling (Fig. 4B). To assess whether HO-1 contributes to the NO-suppressive effect of WSGLR under inflammatory challenge, NO production was measured in LPS-stimulated SIM-A9 cells in the absence or presence of ZnPP (HO-1 inhibitor). Without ZnPP, WSGLR (200 ㎍/mL) substantially reduced LPS-induced NO production; however, in the presence of ZnPP, WSGLR produced only a minimal reduction in NO levels under the same conditions (Fig. 4C).
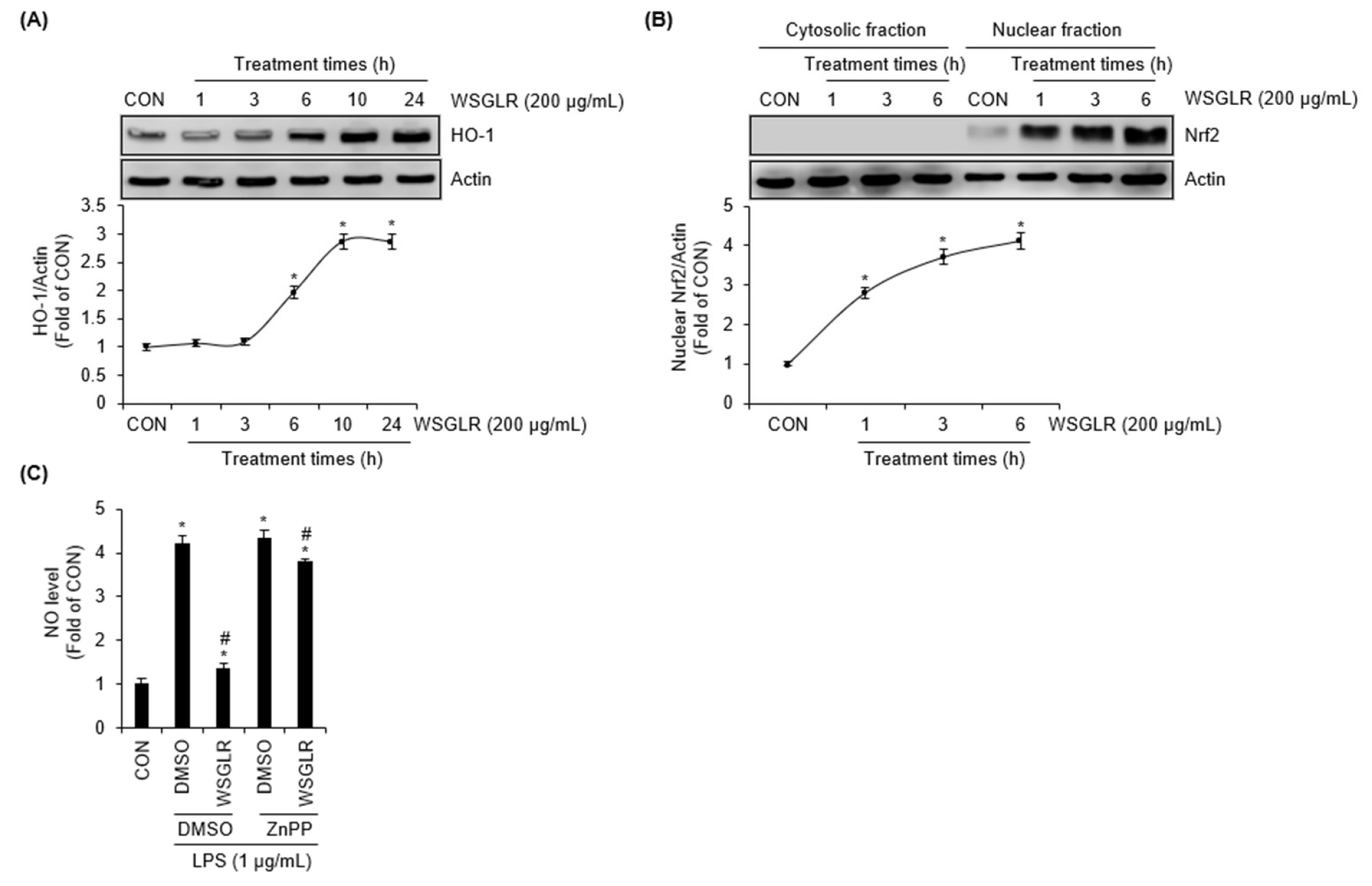
Fig. 4.
WSGLR induces nuclear Nrf2 accumulation and HO-1 expression, and HO-1 inhibition attenuates WSGLR-mediated suppression of NO production in SIM-A9 microglia. (A) SIM-A9 microglial cells were treated with WSGLR (200 ㎍/mL) for the indicated times (1-24 h). Total cell lysates were subjected to immunoblotting to determine HO-1 protein expression. (B) SIM-A9 cells were exposed to WSGLR (200 ㎍/mL) for 1, 3, and 6 h. Cytosolic fractions and Nuclear fractions were prepared and analyzed by immunoblotting for Nrf2. (C) Cells were pretreated with WSGLR (200 ㎍/mL) in the absence or presence of ZnPP (20 μM, HO-1 inhibitor), followed by LPS stimulation. NO production in culture supernatants was quantified by the Griess assay. Data are presented as mean ± SEM from three independent experiments. *p < 0.05 vs. CON, #P < 0.05 vs. LPS-only (DMSO).
PI3K activity contributes to WSGLR-induced Nrf2 nuclear accumulation and HO-1 upregulation in SIM-A9 microglia
Given that WSGLR increased nuclear Nrf2 and HO-1 levels and that HO-1 activity was functionally required for NO suppression under LPS challenge (Fig. 4), we next examined whether PI3K signaling is involved in WSGLR-mediated activation of the Nrf2/HO-1 axis (Fig. 5). In the absence of the PI3K inhibitor LY294002, treatment with WSGLR (200 ㎍/mL) markedly increased HO-1 protein levels and nuclear Nrf2 accumulation in SIM-A9 cells (Fig. 5A and 5B). However, when cells were pretreated with LY294002, the WSGLR-induced increases in HO-1 and nuclear Nrf2 were substantially attenuated compared with the corresponding WSGLR condition without inhibitor (Fig. 5A and 5B). To determine whether WSGLR directly activates PI3K signaling, phosphorylation of PI3K was assessed over time. WSGLR treatment (200 ㎍/mL) induced a rapid increase in the p-PI3K/PI3K ratio, which was noticeably elevated at 1 h and then gradually declined at subsequent time points (3-24 h) (Fig. 5C). Together, these data indicate that early PI3K activation by WSGLR is associated with, and is required for full induction of, the downstream Nrf2 nuclear response and HO-1 upregulation in SIM-A9 microglia.
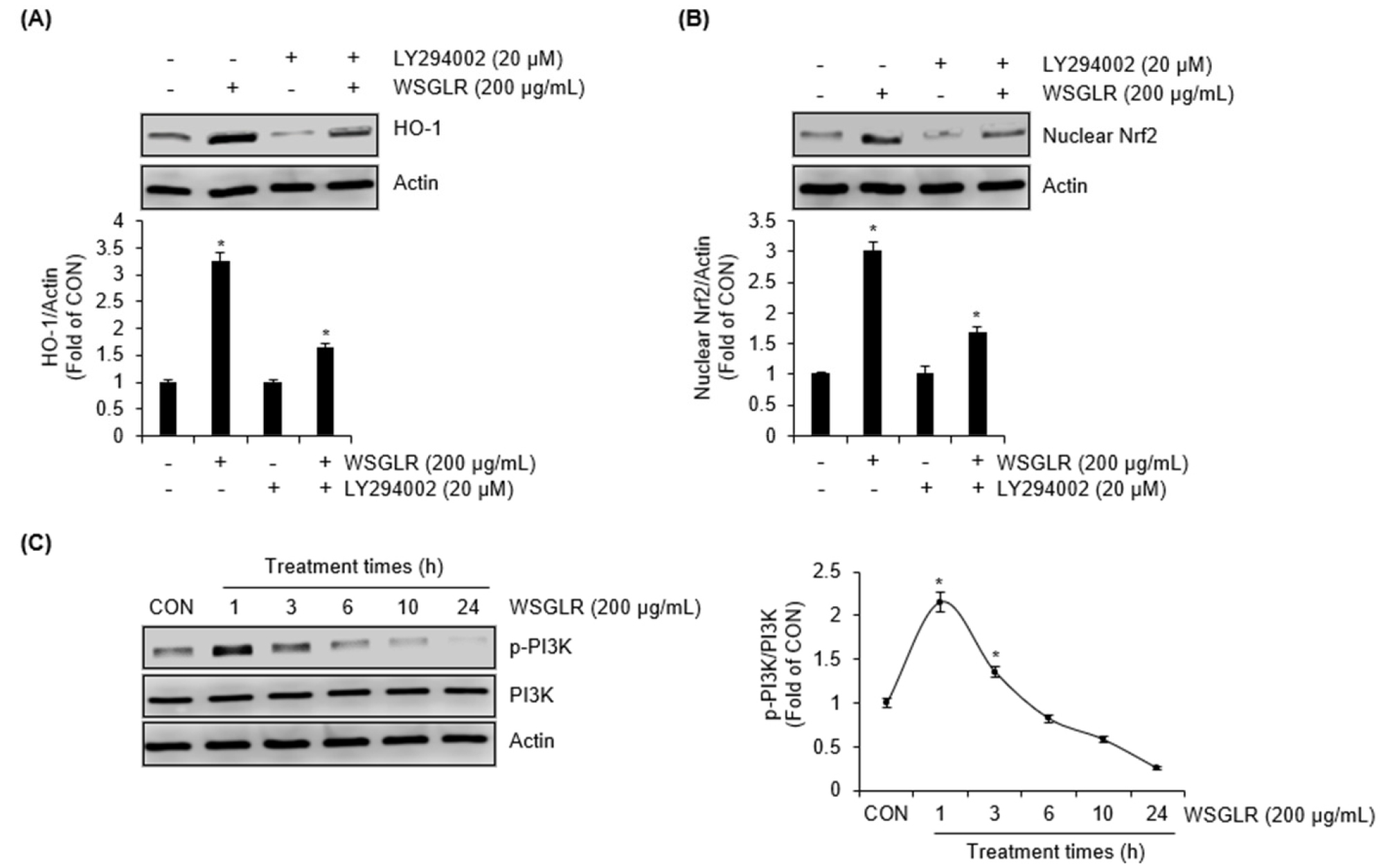
Fig. 5.
PI3K signaling is required for WSGLR-mediated activation of the Nrf2/HO-1 axis in SIM-A9 microglia. (A) SIM-A9 cells were pretreated with the PI3K inhibitor LY294002 (20 μM) or vehicle for 2 h, followed by exposure to WSGLR (200 ㎍/mL) for 6 h. Total cell lysates were subjected to immunoblotting to determine HO-1 protein expression. (B) SIM-A9 cells were pretreated with the PI3K inhibitor LY294002 (20 μM) or vehicle for 2 h, followed by exposure to WSGLR (200 ㎍/mL) for 1 h. Nuclear fractions were subjected to immunoblotting to determine the Nrf2 protein level. (C) To evaluate direct activation of PI3K signaling by WSGLR, SIM-A9 cells were treated with WSGLR (200 ㎍/mL) for the indicated times (1-24 h). Phosphorylation of PI3K was analyzed by immunoblotting, and the p-PI3K/PI3K ratio was quantified. Data are presented as mean ± SEM from the three independent experiments. *p < 0.05 vs. CON.
WSGLR protects SH-SY5Y cells against Aβ25-35-induced cytotoxicity
After establishing that WSGLR suppresses LPS-driven inflammatory signaling and downstream iNOS/NO output in SIM-A9 microglia, we next evaluated whether WSGLR also confers direct neuroprotection in a neuronal cell model challenged with Aβ25-35 (Fig. 6). In SH-SY5Y cells, exposure to Aβ25-35 alone (DMSO group) markedly reduced cell viability, corresponding to an approximately 45.2% decrease relative to the CON (Fig. 6A). Co-treatment with WSGLR mitigated this loss of viability in a concentration-dependent manner: the Aβ25-35-associated viability reduction was approximately 39.4% at 50 μg/mL, 20.2% at 100 ㎍/mL, and 7.7% at 200 ㎍/mL compared with the CON (Fig. 6A). Consistent with these viability results, Aβ25-35 alone increased LDH release to approximately 162.3% above the CON level (Fig. 6B). WSGLR co-treatment reduced LDH release under Aβ25-35 challenge, with LDH increases of approximately 154.7%, 128.7%, and 110.8% above the CON at 50, 100, and 200 ㎍/mL, respectively (Fig. 6B). To further assess whether WSGLR modulates apoptosis-related protein changes triggered by Aβ25-35, we examined cleaved PARP, Bax, and Bcl-2 levels (Fig. 6C). Aβ25-35 alone increased cleaved PARP and Bax and decreased Bcl-2 compared with the CON. In contrast, WSGLR at 100 and 200 ㎍/mL attenuated the Aβ25-35-induced increases in cleaved PARP and Bax and alleviated the decrease in Bcl-2 (Fig. 6C). Finally, intracellular ROS generation was quantified to determine whether WSGLR influences Aβ25-35-associated oxidative stress (Fig. 6D). Aβ25-35 alone increased ROS to approximately 2.6-fold relative to the CON. WSGLR reduced ROS accumulation under Aβ25-35 challenge, with ROS levels of approximately 1.96-fold at 100 ㎍/mL and 1.24-fold at 200 ㎍/mL relative to the CON (Fig. 6D).
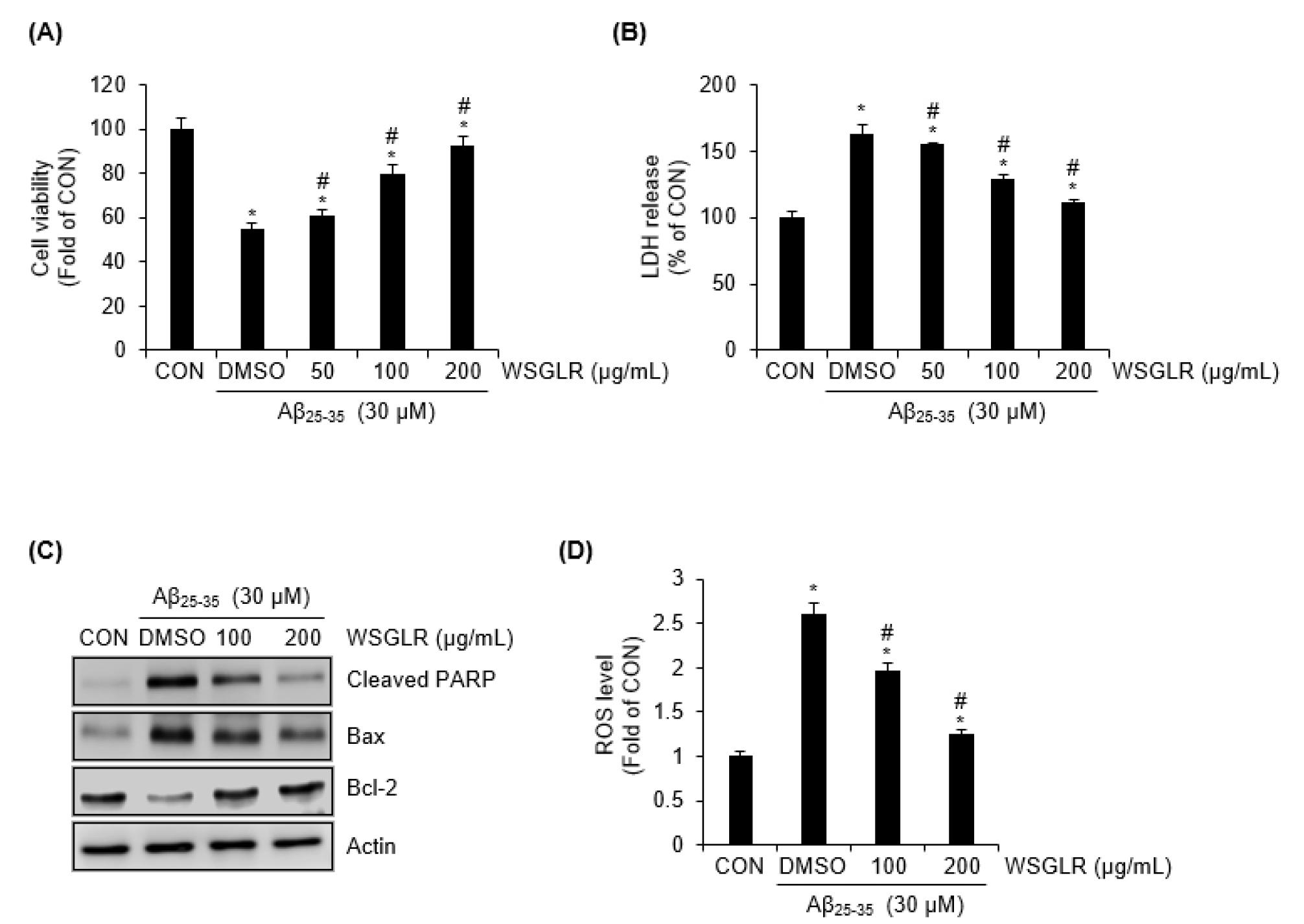
Fig. 6.
WSGLR protects SH-SY5Y neuronal cells against Aβ25-35-induced cytotoxicity, apoptosis-related protein changes, and oxidative stress. (A) SH-SY5Y cells were exposed to Aβ25-35 in the absence or presence of WSGLR (50, 100, or 200 ㎍/mL). Cell viability was determined using an MTT assay and expressed relative to the untreated control (CON). (B) Cytotoxicity was evaluated by measuring lactate dehydrogenase (LDH) release in culture supernatants under the same treatment conditions as in (A). Data are presented as percent change relative to CON. (C) To assess apoptosis-related protein modulation, total cell lysates were analyzed by immunoblotting for cleaved PARP, Bax, and Bcl-2 following treatment with Aβ25-35with or without WSGLR (100 and 200 ㎍/mL). (D) Intracellular ROS generation was quantified using a fluorescent ROS probe after Aβ25-35 exposure in the absence or presence of WSGLR (100 and 200 ㎍/mL). ROS levels are expressed as fold change relative to CON. Data are presented as mean ± SEM from three independent experiments. *p < 0.05 vs. CON, #P < 0.05 vs. Aβ25-35-only (DMSO).
Discussion
Neuroinflammation and oxidative stress are widely recognized as convergent drivers of neurodegeneration, including Alzheimer’s disease (AD), where microglial activation and amyloid-associated neuronal injury mutually reinforce disease progression (Cai et al., 2022; Heneka et al., 2015). In this study, we investigated a wild-simulated ginseng blended formulation (WSGLR, WSGL:WSGR = 1:1, w/w) and demonstrated two complementary outcomes such as suppression of LPS-driven inflammatory output in microglia and direct protection of neuronal cells against Aβ25-35-induced cytotoxicity. The data collectively support a model in which WSGLR down-modulates pro-inflammatory signaling (p38/JNK and NF-κB) while engaging a PI3K-linked Nrf2/HO-1 cytoprotective program, ultimately converging on reduced iNOS/NO generation in microglia and attenuated oxidative/apoptotic injury in neuronal cells.
A central design feature of this work is the use of a defined-ratio blend of aerial and underground extracts. Ginsenoside composition is known to be strongly tissue-dependent in Panax species, with leaves/stems often showing ginsenoside patterns and abundances that differ from the main root (Kang and Kim, 2016). In addition, stem-leaf tissues have been proposed as a rich and sustainable source of ginsenosides and related saponins, providing a scientific rationale for incorporating aerial tissues rather than restricting formulations to roots alone (Zhang et al., 2021). Consistent with this concept, our targeted profiling revealed clear part-specific ginsenoside signatures (e.g., F3/F5/Rd2 detected only in WSGL, and Ra1-Ra3 detected only in WSGR), indicating that the two extracts contribute non-overlapping chemical features. This compositional complementarity provides a phytochemical justification for testing WSGLR as a blended formulation rather than treating WSGL and WSGR as interchangeable sources.
To assess microglial inflammatory responses, we used SIM-A9 cells, a spontaneously immortalized murine microglial line that retains key microglial characteristics and responds robustly to inflammatory stimuli such as LPS, including induction of nitric oxide production (Nagamoto-Combs et al., 2014). In the initial screening, WSGLR reduced LPS-induced NO output more strongly than either WSGL or WSGR alone in the same test condition. While these data do not by themselves establish synergy, they indicate that combining part-specific phytochemical profiles can translate into improved functional suppression of a canonical microglial inflammatory readout. The concentration-response experiments further strengthen this conclusion by showing that WSGLR reduced both NO production and iNOS expression in a dose-dependent manner without detectable cytotoxicity within the tested range. The signaling analysis provides mechanistic coherence: WSGLR attenuated LPS-induced phosphorylation of p38 and JNK and reduced NF-κB p65 activation, whereas ERK1/2 phosphorylation was not noticeably altered. This pattern is consistent with the established role of p38/JNK and NF-κB in microglial pro-inflammatory gene induction, including iNOS-dependent NO production under endotoxin challenge (Kang et al., 2013; Lee et al., 2025). Notably, selective modulation of MAPKs (rather than uniform suppression of all MAPKs) is commonly observed with natural products and saponin-rich preparations and can reflect pathway- or node-specific regulation rather than nonspecific cytotoxicity. From a phytochemical perspective, the prominent ginsenosides in WSGL and WSGR (e.g., Re/Rg1/Rd in WSGL and Ra2/Rb1 in WSGR) are all within classes previously associated with immunomodulatory or anti-inflammatory actions in cellular systems. For instance, ginsenoside Re has been reported to reduce NO formation and suppress NF-κB-related inflammatory signaling in microglial models, supporting the plausibility that ginsenoside-rich fractions can target inflammatory outputs relevant to our readouts (Lee et al., 2012). Likewise, ginsenoside Rg1 has been reported to inhibit LPS-associated inflammatory signaling through NF-κB/MAPK-related pathways (Hu et al., 2011). Although the present work does not assign causality to any single ginsenoside, the observed part-specific profiles and the improved functional performance of the blend support the idea that WSGLR provides broader coverage of bioactive saponins than either extract alone.
In parallel with suppression of pro-inflammatory signaling, WSGLR activated a cytoprotective pathway characterized by early nuclear accumulation of Nrf2 followed by delayed induction of HO-1 protein. This temporal sequence is biologically plausible because Nrf2 nuclear translocation is an upstream event that precedes transcriptional induction of phase II/antioxidant enzymes, including HO-1. Moreover, the functional inhibitor experiment strengthens inference beyond correlation: inhibition of HO-1 with ZnPP markedly diminished the NO-suppressive effect of WSGLR under LPS challenge, indicating that HO-1 activity contributes materially to suppression of inflammatory output in this system. The concept that Nrf2-dependent HO-1 induction counter-regulates microglial NO/iNOS output has precedent in microglial inflammation models, in which HO-1 induction is linked to reduced NO production and inflammatory signaling (Kang et al., 2013). Our PI3K-related data further supports an ordered signaling cascade. WSGLR rapidly increased p-PI3K/PI3K at early time points, and pharmacological PI3K inhibition with LY294002 attenuated WSGLR-induced nuclear Nrf2 accumulation and HO-1 upregulation. PI3K/Akt signaling is frequently reported as an upstream regulator of Nrf2 activation and HO-1 expression in diverse cellular contexts, and LY294002 is widely used to functionally probe PI3K dependence in Nrf2/HO-1 induction paradigms (Xu et al., 2015). Taken together, these results are consistent with a mechanism in which WSGLR triggers early PI3K activation that supports robust Nrf2 nuclear response and subsequent HO-1 induction, thereby contributing to suppression of iNOS/NO output under inflammatory stimulation. Importantly, our data do not require the assumption that the PI3K/Nrf2/HO-1 axis is the sole driver of the anti-inflammatory phenotype, because WSGLR also dampened p38/JNK and NF-κB activation. A parsimonious interpretation is that WSGLR modulates LPS responses through complementary mechanisms: attenuation of pro-inflammatory MAPK/NF-κB signaling and activation of cytoprotective PI3K/Nrf2/HO-1 signaling, both converging on reduced NO production.
Beyond anti-neuroinflammatory actions in microglia, WSGLR provided direct neuroprotection in SH-SY5Y cells challenged with Aβ25-35, a commonly used peptide fragment model to induce amyloid-associated cytotoxicity, oxidative stress, and apoptosis-like changes in neuronal cells (Kozina et al., 2024). The protective profile was internally consistent across independent endpoints: WSGLR improved cell viability, reduced LDH release (membrane damage), attenuated Aβ-induced shifts in apoptosis-related proteins (reduced cleaved PARP and Bax, restored Bcl-2), and decreased ROS accumulation. Similar multi-parameter protective patterns spanning oxidative stress and apoptotic signaling are frequently used to define neuroprotective effects in Aβ25-35-stimulated SH-SY5Y systems (He et al., 2023). These neuronal results align with prior observations that ginseng-derived preparations can mitigate Aβ25-35 toxicity through regulation of oxidative stress and apoptosis-related pathways. For example, red ginseng oil and/or related ginseng preparations have been reported to protect against Aβ25-35-induced toxicity by modulating ROS and apoptosis-associated mechanisms (Lee et al., 2017; Lee et al., 2018). Our data extend the concept to a defined-ratio aerial-underground blended extract, and link microglial anti-inflammatory mechanisms with direct neuronal protective effects within the same study.
In summary, WSGLR exhibited robust anti-neuroinflammatory activity in microglia and direct neuroprotective activity in neuronal cells. Mechanistically, WSGLR attenuated LPS-induced p38/JNK and NF-κB activation, engaged PI3K-dependent Nrf2 nuclear accumulation and HO-1 induction, and required HO-1 activity for full suppression of inflammatory NO output. In neuronal cells, WSGLR alleviated Aβ25-35-induced cytotoxicity, membrane damage, oxidative stress, and apoptosis-associated signaling. Together, these findings support WSGLR as a blended extract with dual anti-neuroinflammatory and neuroprotective potential, warranting further validation in advanced translational models.
