

Introduction
Materials and Methods
Chemical reagents
Preparation of 11S,17S-DiHDHA
High-performance liquid chromatography (HPLC) analysis
Cell culture
Griess assay
Reverse transcription polymerase chain reaction (RT-PCR)
Transfection of small interference RNA (siRNA)
Nuclear protein extraction
SDS-PAGE and Western blot analysis
Measurement of cell viability
Measurement of LDH release
Measurement of the intracellular ROS level
Statistical analysis
Results
Biotransformation and purification of 11S,17S-DiHDHA
11S,17S-DiHDHA inhibits NO production through suppressing iNOS expression in LPS-stimulated SIM-A9 cells
11S,17S-DiHDHA inhibits MAPK and NF-κB signaling pathway in LPS-stimulated SIM-A9 cells
11S,17S-DiHDHA activates Nrf2/HO-1 signaling pathway in SIM-A9 cells
11S,17S-DiHDHA-mediated activation of Nrf2/HO-1 signaling pathway is dependent on PI3K activation in SIM-A9 cells
11S,17S-DiHDHA inhibits Aβ25-35-induced cell death in SH-SY5Y cells
Discussion
Introduction
Alzheimer’s disease (AD), a neurodegenerative disorder that disrupts learning and memory processes, is an irreversible brain disorder and the most prevalent form of dementia (Zvěřová, 2019). The prevalence of AD has been rising at an alarming rate each year, and it has been reported that without the development of novel preventive or therapeutic strategies, the number of AD patients is projected to exceed 100 million by 2050 (Querfurth and LaFerla, 2010).
Neuroinflammation, characterized by the activation of glial cells such as microglia and astrocytes, is recognized as a key mechanism for maintaining brain homeostasis (Sobue et al., 2023). However, excessive neuroinflammatory responses have been reported to accelerate the pathogenic progression of AD (Heneka et al., 2015). Excessive inflammatory responses induce microglia to release substantial amounts of pro-inflammatory mediators, which contribute to neurotoxicity by promoting oxidative stress and neuronal apoptosis (Chen et al., 2024). This process is recognized as one of the major pathological triggers of AD (Chen et al., 2024). Moreover, an appropriate neuroinflammatory response by microglia facilitates the effective clearance of excessive β-amyloid (Aβ) through phagocytosis (Brown et al., 2020). However, excessive microglial activation can exacerbate Aβ aggregation, thereby accelerating the progression of AD (Brown et al., 2020). The excessive accumulation of Aβ is known to create a deleterious feedback loop that accelerates neurodegeneration and neuronal apoptosis (Weekman et al., 2014). This pathological cycle further impairs microglial phagocytosis, thereby diminishing Aβ clearance and exacerbating its accumulation, ultimately contributing to the progression of AD (Weekman et al., 2014). Furthermore, Aβ accumulation has been reported to induce neurotoxicity through oxidative stress mediated by reactive oxygen species (ROS) (Muthaiyah et al., 2011). Thus, reducing or inhibiting excessive neuroinflammatory responses and Aβ-mediated neurotoxicity could serve as promising strategies for the prevention and treatment of AD.
Specialized pro-resolving mediators (SPMs), recognized for their potent anti-inflammatory activity, have been reported to cross the blood-brain barrier and attenuate excessive microglial neuroinflammation (Ponce et al., 2022). By modulating the overproduction of pro-inflammatory mediators, SPMs such as RvD2, LXA4, and AnxA1 contribute to neuroprotection in AD (Ponce et al., 2022; Valente et al., 2022). Protectin D1 (PD1), also known as 10R,17S-dihydroxydocosahexaenoic acid (10R,17S-DiHDHA), has been reported to exhibit anti-inflammatory (Iwuchukwu et al., 2022), anti-apoptotic (Antony et al., 2010), and neuroprotective activities (Asatryan and Bazan, 2017; Belayev et al., 2018). Protectin Dx (PDX, 10S,17S-DiHDHA), which exhibits structural differences from protectin D1 (PD1), has been reported to possess anti-inflammatory (Lagarde et al., 2020), antioxidant (Hwang et al., 2019), and antiviral activities (Lagarde et al., 2020). However, unlike PD1, PDX has not been shown to confer neuroprotective effects, suggesting that even within the same class of SPMs, structural differences can lead to distinct pharmacological functionalities. 11S,17S-DiHDHA is a SPM that represents the 11-isomer form of PDX. However, to date, no studies have investigated its pharmacological properties, leaving its potential biological functions largely unexplored. Therefore, in this study, we aim to demonstrate the neuroprotective activity of 11S,17S-DiHDHA by showing its ability to suppress neuroinflammation in SIM-A9 cells and inhibit Aβ-mediated cell death in SH-SY5Y cells.
Materials and Methods
Chemical reagents
Docosahexaenoic acid (DHA, cat. no. D2534) and L-cysteine (cat. no. C0515) were purchased from Sigma-Aldrich (St. Louis, MO, USA) and Tokyo Chemical Industry (Tokyo, Japan), respectively. 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT, cat. no. 475989), LY294002 (cat. no. 440202), Griess reagent (cat. no. G4410), and lipopolysaccharide (LPS, cat. no. L2630), and Aβ25-35 (cat. no. A4559) were procured from Sigma-Aldrich (St. Louis, MO, USA). Zinc (II) Protoporphyrin IX (ZnPP, cat. no. ALX-430-049-M025) was purchased from Enzo Life Sciences (Long Island, NY, USA). Primary antibodies such as extracellular signal-regulated protein kinase 1 and 2 (ERK1/2; cat. no. 9102), and phosphorylated (p-)ERK1/2 (cat. no: 4377), p38 (cat. no: 9212), p-p38 (cat. no. 4511), and c-Jun N-terminal kinase (JNK, cat. no. 9258), and p-JNK (cat. no: 9251), p65 (cat. no: 8242), p-p65 (cat. no. 3033), and phosphoinositide 3-kinase (PI3K; cat. no. 4257), and p-PI3K (cat. no. 4228) and heme oxygenase 1 (HO-1; cat. no. 70081), nuclear factor erythroid 2-related factor 2 (Nrf2, cat. no. 12721), and β-actin (cat. no. 5125) were purchased from Cell Signaling Technology (Danvers, MA, USA). Primary antibodies such as Bcl-2 (cat. no. sc-7382) and Bax (cat. no. sc-7480) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Secondary antibodies for anti-rabbit IgG, HRP-linked antibody (cat. no. 7074) and Anti-mouse IgG, HRP-linked antibody (cat. no. 7076) were purchased from Cell Signaling Technology (Danvers, MA, USA). Additionally, control siRNA (cat. no. 6568) and Nrf2 siRNA (cat. no. sc-37049) were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA) and Santa Cruz Biotechnology, Inc. (Dallas, TX, USA), respectively.
Preparation of 11S,17S-DiHDHA
11S,17S-Dihydroxydocosahexaenoic acid (11S,17S-DiHDHA) was synthesized from DHA using recombinant Escherichia coli expressing a double-oxygenating 9S-lipoxygenase from Sphingopyxis macrogoltabida (Oh et al., 2022). The reactions were performed at 35°C in 50 mM N-cyclohexyl-2-aminoethanesulfonic acid (CHES) buffer (pH 8.5) containing 6 mM DHA as a substrate and 4.0 g/L of cells with 200 mM cysteine as a reducing agent for 60 min. The 11S,17S-DiHDHA product was extracted by adding an equal volume of ethyl acetate to the reaction mixture. The solvent was then removed by evaporation under reduced pressure until the solution was completely dry. After drying, the residue was resuspended in an equal volume of methanol. The methanol solution was subjected to a preparative high-performance liquid chromatography (HPLC) on an Agilent 1260 system (Santa Clara, CA, USA) equipped with a Nucleosil C18 preparative column (10 × 250 ㎜; Phenomenex, Torrance, CA, USA) at 30°C with a flow rate of 6 mL/min. The column was eluted with a gradient of solvent A (acetonitrile/water/acetic acid, 50:50:0.1, v/v/v) and solvent B (acetonitrile/water/acetic acid, 100:0:0.1, v/v/v) as follows: 100:0 for 5 min, a gradient from 100:0 to 0:100 from 5 to 21 min, 0:100 from 21 to 27 min, a gradient from 0:100 to 100:0 from 27 to 32 min, and 100:0 from 32 to 35 min. Absorbance was monitored at 202 nm to detect the 11S,17S-DiHDHA product, and the product fractions were collected based on the peaks observed in the chromatogram. The purity of the isolated samples was greater than 90%.
High-performance liquid chromatography (HPLC) analysis
After the reactions, the reaction mixtures were extracted with an equal volume of ethyl acetate. The extracts were dried and resuspended in methanol for HPLC analysis (Agilent 1260). The concentrations of DHA and 11S,17S-DiHDHA in methanol solutions were quantified using HPLC at 202 ㎚ on a Nucleosil-C18 column. The column was eluted at 30°C with a flow rate of 0.25 mL/min, using a gradient from solvent A to solvent B as follows: 100:0 for 5 min, a gradient from 100:0 to 0:100 from 5 to 15 min, 0:100 from 15 to 20 min, and a gradient from 0:100 to 100:0 from 20 to 25 min.
Cell culture
A microglial cell line, SIM-A9 cells (cat. no. CRL-3265) and a neuroblastoma cell line, SH-SY5Y cells (cat. no. CRL-2266) were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). SIM-A9 and SH-SY5Y cells were maintained in a CO2 incubator at 37°C with 5% CO2, using DMEM/F-12 medium supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 ㎍/mL streptomycin.
Griess assay
The inhibitory effect of 11S,17S-DiHDHA on LPS-induced nitric oxide (NO) production in SIM-A9 cells was evaluated using the Griess assay. When SIM-A9 cells cultured in a 12-well plate reached approximately 90% confluence, they were pretreated with 11S,17S-DiHDHA for 2 h followed by stimulation with LPS at a concentration of 1 ㎍/mL for 18 h. Subsequently, the cell culture supernatant was mixed with Griess reagent at a 1:1 ratio (v/v) and incubated at room temperature for 15 min. Absorbance was measured at 540 ㎚ using a UV/visible spectrophotometer (SpectraMax M2, Molecular Devices, San Jose, CA, USA).
Reverse transcription polymerase chain reaction (RT-PCR)
The inhibitory effect of 11S,17S-DiHDHA on LPS-induced iNOS overexpression in SIM-A9 cells was evaluated using RT-PCR. When SIM-A9 cells in a 6-well plate reached approximately 90% confluence, they were pretreated with 11S,17S-DiHDHA for 2 h, followed by LPS stimulation at 1 ㎍/mL for 18 h. After 18 h, total RNA was extracted from SIM-A9 cells using the RNeasy Mini Kit (Qiagen, Hilden, Germany). Following quantification of total RNA, 1 ㎍ of total RNA was used for cDNA synthesis using the Verso cDNA Kit (Thermo Fisher Scientific, Waltham, MA, USA). After cDNA synthesis, the cDNA was subsequently amplified by PCR using the PCR Master Mix Kit (Promega, Madison, WI, USA) and iNOS primers. The primer sequences used for iNOS and GAPDH in this study were as follows: A and B. iNOS mRNA, F: 5′- GTTACCATGAGGCTGAAATCC-3′ and R: 5′-CCTCTTGTCTTTGACCCAGTAC-3′, GAPDH mRNA, F: 5′-GGACCTCATGGCCTACATGG-3′ and R: 5′- TAGGGCCTCTCTTGCTCAGT-3′. Following PCR amplification, the resulting PCR products were visualized using agarose gel electrophoresis, and the intensity of the visualized mRNA bands was quantified using UN-SCAN-IT gel software version 5.1 (Silk Scientific Inc., Vineyard, UT, USA).
Transfection of small interference RNA (siRNA)
SIM-A9 were plated in 6-well plates and incubated overnight to allow proper adhesion. Once attached, the cells were transfected with either a non-targeting control siRNA or Nrf2-specific siRNA at a final concentration of 100 nM. The transfection procedure was carried out over a 48 hour period using the TransIT-TKO transfection reagent (Mirus, Madison, WI, USA), following the manufacturer’s protocol.
Nuclear protein extraction
After completing all treatments, nuclear proteins were extracted from SIM-A9 cells using the Nuclear Extract Kit (Active Motif, Carlsbad, CA, USA) according to the manufacturer’s instructions. The extracted nuclear proteins were stored at -80°C until further analysis.
SDS-PAGE and Western blot analysis
Following the completion of all treatments, the concentrations of the cellular protein extracted using RIPA Lysis and Extraction Buffer (Thermo Fisher Scientific, Waltham, MA, USA) were determined using a Pierce™ BCA Protein Assay Kits (Thermo Fisher Scientific, Waltham, MA, USA). Proteins were then resolved by SDS-PAGE under conditions of 150 V and 400 A for 1 h, followed by electrotransfer onto a nitrocellulose membrane (Thermo Fisher Scientific, Waltham, MA, USA) at 100 V and 300 A for 2 h To minimize nonspecific binding, membranes were blocked with 5% non-fat milk at room temperature for 1 h. Subsequently, they were incubated overnight at 4°C with primary antibodies diluted 1:1,000, followed by a 1-hour incubation at room temperature with a secondary antibody at the same dilution. Protein bands were detected using a LI-COR C-DiGit Blot Scanner (LI-COR Biosciences, Lincoln, NE, USA) after treatment with ECL Prime Western Blotting Detection Reagents (Amersham Biosciences Corp., Piscataway, NJ, USA). Protein nand intensities were quantified using UN-SCAN-IT gel software version 5.1 (Silk Scientific Inc., Vineyard, UT, USA).
Measurement of cell viability
The inhibitory effect of 11S,17S-DiHDHA on Aβ25-35-induced reduction in the viability of SH-SY5Y cells was evaluated using the MTT assay. When SH-SY5Y cells reached approximately 90% confluence in a 96-well plate, they were pretreated with 11S,17S-DiHDHA for 2 h, followed by Aβ25-35 treatment, and then incubated for an additional 24 h. After 24 hours of treatment, the culture medium was discarded and replaced with MTT solution at a final concentration of 0.5 ㎎/mL. The cells were then incubated at 37°C with 5% CO2 for 4 h to allow for formazan crystal formation. Following incubation, the MTT solution was carefully removed, and 100 μL of DMSO was added to dissolve the resulting insoluble formazan. The absorbance was measured at 570 ㎚ using a UV/visible spectrophotometer (SpectraMax M2, Molecular Devices, San Jose, CA, USA).
Measurement of LDH release
When SH-SY5Y cells reached approximately 90% confluence in a 12-well plate, they were pretreated with 11S,17S-DiHDHA for 2 h, followed by Aβ25-35 treatment, and then incubated for an additional 24 h. After 24 hours of treatment, the amount of LDH released into the culture medium was measured using the CyQUANT™ LDH Cytotoxicity Assay Kit (Invitrogen Co., Carlsbad, CA, USA) according to the manufacturer’s protocol.
Measurement of the intracellular ROS level
When SH-SY5Y cells reached approximately 90% confluence in a 12-well plate, they were pretreated with 11S,17S-DiHDHA for 2 h, followed by Aβ25-35 treatment, and then incubated for an additional 24 h. After 24 hours of treatment, the amount of LDH released into the culture medium was measured using the Intracellular ROS Assay Kit (CELL BIOLABS, INC., San Diego, CA, USA) according to the manufacturer’s protocol.
Statistical analysis
All experiments were repeated at least three times. Statistical analysis was performed using GraphPad Prism version 5.0 (GraphPad Software, Inc.) and data are presented as the mean ± standard deviation. Data were analyzed using one-way analysis of variance followed by Bonferroni’s post hoc test. P<0.05 was considered to indicate a statistically significant difference.
Results
Biotransformation and purification of 11S,17S-DiHDHA
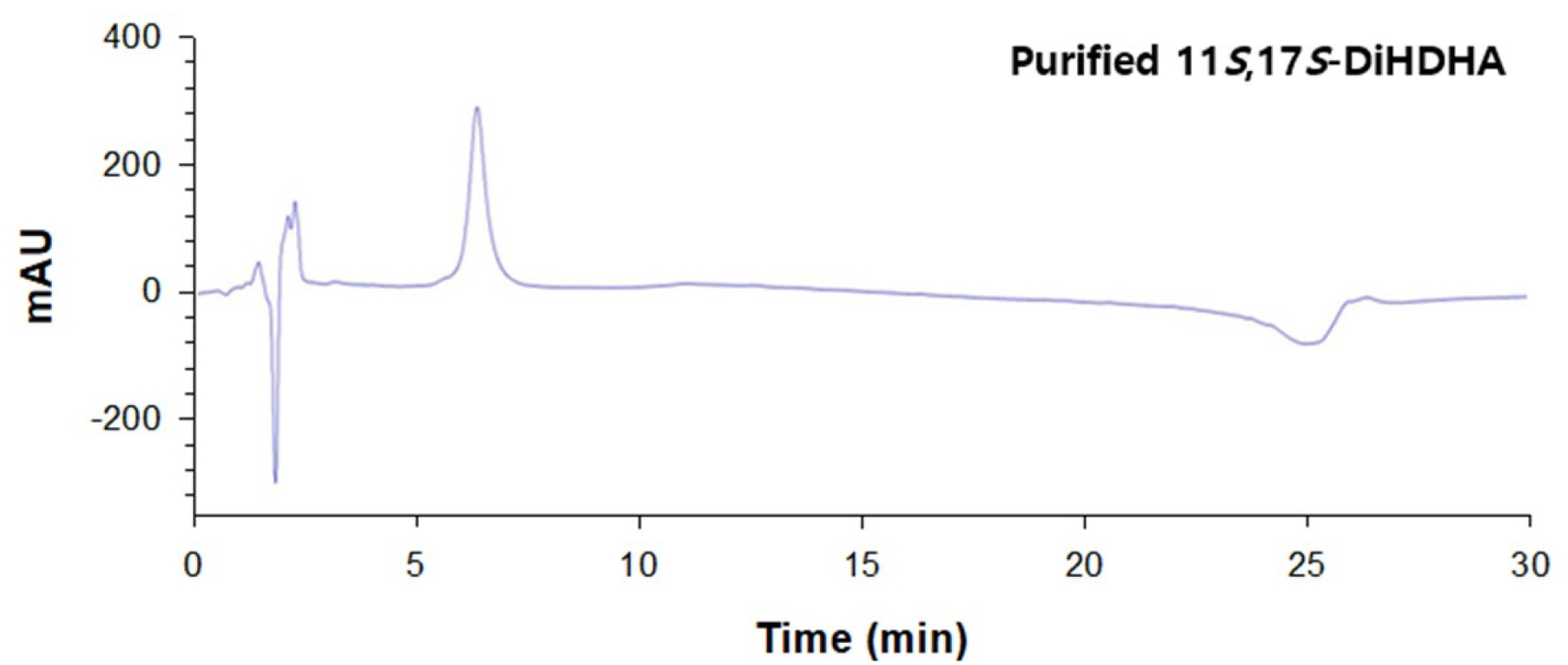
The biotransformation to produce 11S,17S-DiHDHA was performed using recombinant E. coli expressing SM 9S-LOX with DHA as substrates in the presence of cysteine. The recombinant E. coli expressing SM 9S-LOX converted 6 mM DHA to 4.1 mM 11S,17S-DiHDHA in 60 min (Fig. 1). The biotransformed products were subsequently purified as DiHDHA with a purity of >93% by Prep-HPLC preparation (Fig. 2).
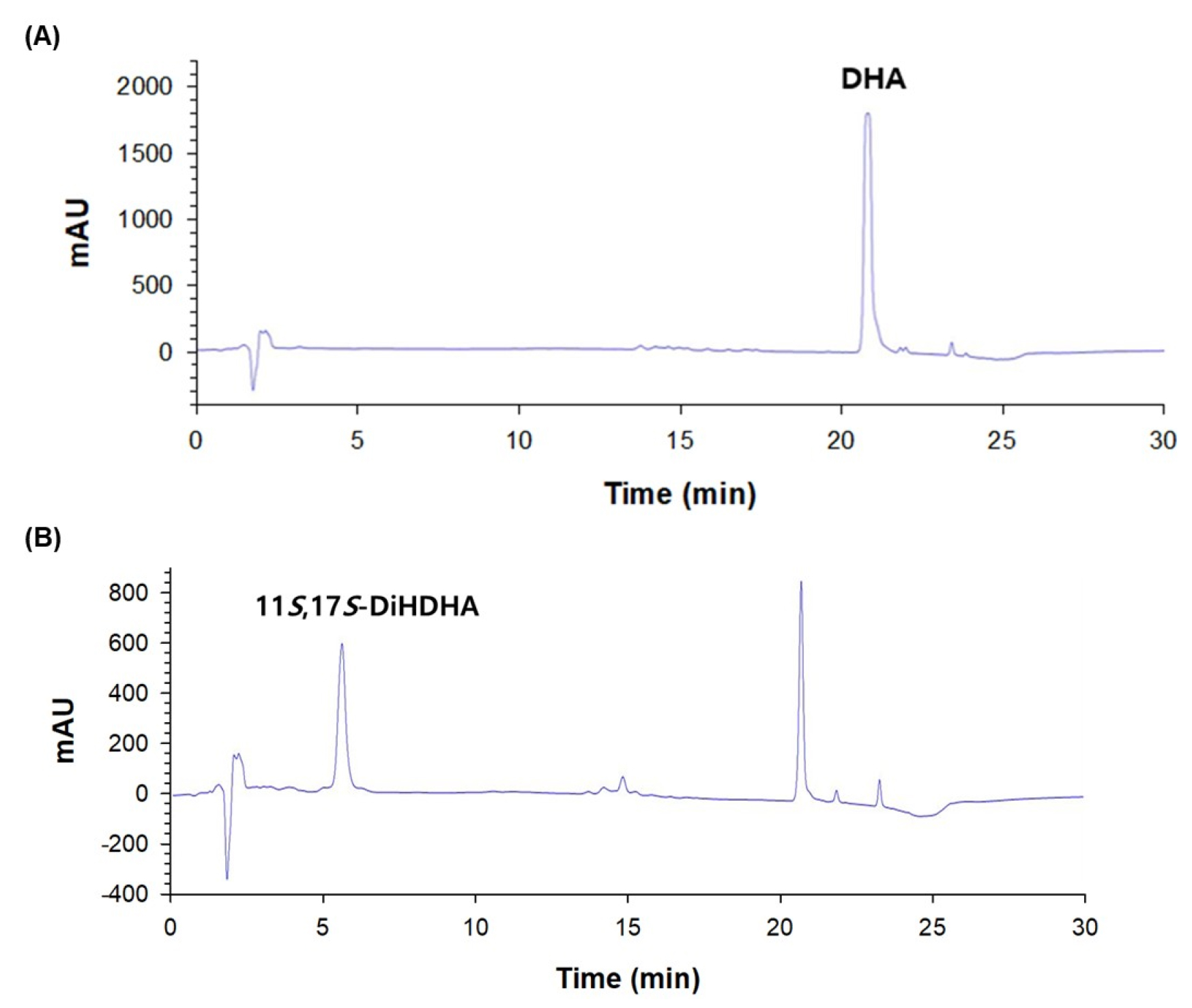
Fig. 1.
HPLC chromatograms with reversed-phase column of DHA standard and 11S,17S-DiHDHA (PDX 11-isomer) obtained from the conversion of DHA by S. macrogoltabida 9S-LOX. (A) HPLC chromatogram of DHA standard. (B) HPLC chromatogram of the product obtained from the conversion of DHA in the presence of cysteine by S. macrogoltabida 9S-LOX.
11S,17S-DiHDHA inhibits NO production through suppressing iNOS expression in LPS-stimulated SIM-A9 cells
To investigate the potential anti-neuroinflammatory effects of 11S,17S-DiHDHA, its ability to suppress NO production in LPS-stimulated SIM-A9 cells was evaluated. As shown in Fig. 3A, compared to the untreated control (CON), LPS treatment alone significantly induced excessive NO production in SIM-A9 cells. However, 11S,17S-DiHDHA markedly suppressed LPS-induced NO overproduction in a statistically significant manner. It is well established that inflammatory stimuli such as LPS activate microglia, leading to increased iNOS expression, which subsequently enhances NO production and release (Chao et al., 1992). To determine whether the 11S,17S-DiHDHA-mediated suppression of NO production was attributed to the inhibition of iNOS expression, we analyzed the changes of iNOS expression by 11S,17S-DiHDHA in LPS-stimulated SIM-A9 cells using RT-PCR. As a result, 11S,17S-DiHDHA effectively suppressed LPS-induced iNOS overexpression in SIM-A9 cells (Fig. 3B). Taken together, the results from Fig. 3A and Fig. 3B indicate that 11S,17S-DiHDHA suppresses NO overproduction in LPS-stimulated SIM-A9 cells by downregulating iNOS expression. This suggests that 11S,17S-DiHDHA possesses anti-neuroinflammatory activity.
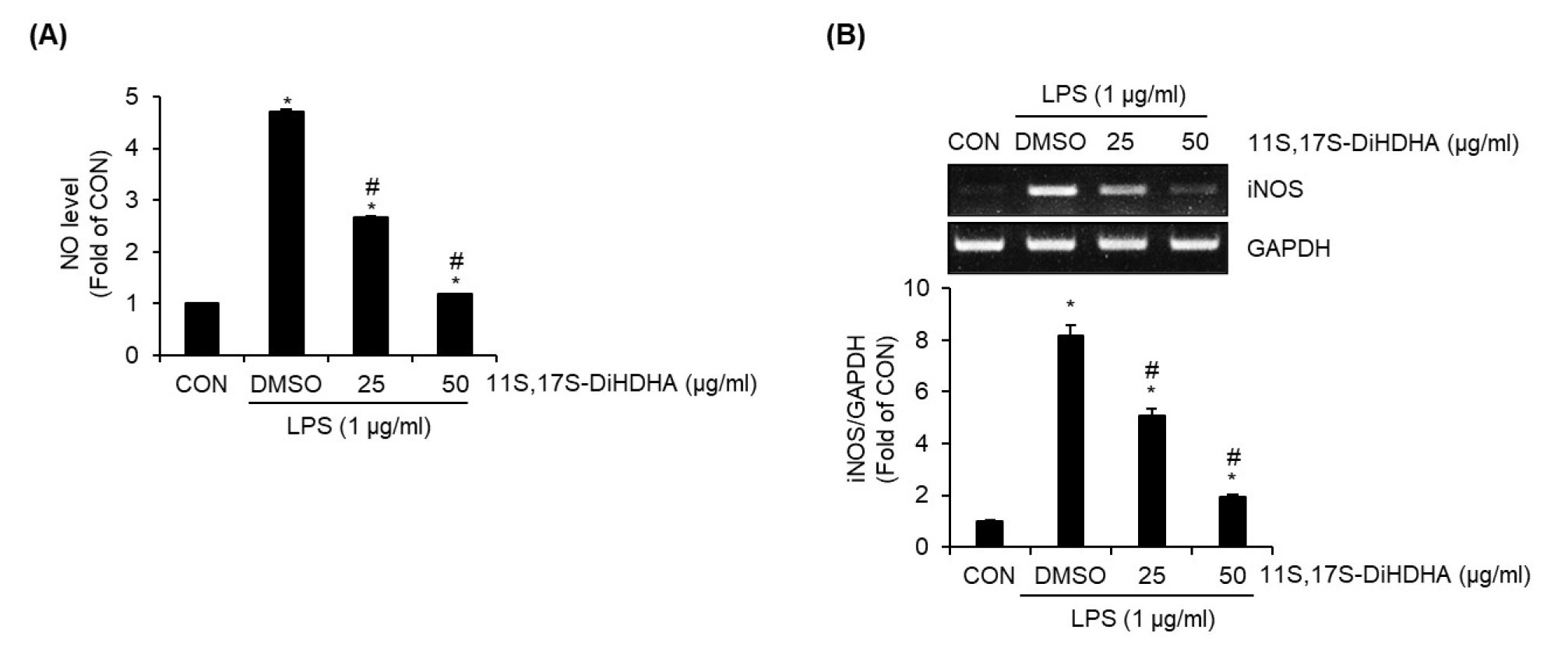
Fig. 3.
Effect of 11S,17S-DiHDHA on the NO production and iNOS expression in LPS-stimulated SIM-A9 cells. SIM-A9 cells were pretreated with 11S,17S-DiHDHA for 2 h and then co-treated with LPS for 18 h. NO level (A) and iNOS expression level (B) were measured using Griess assay and RT-PCR. *P < 0.05 vs CON (untreated group). #P < 0.05 vs DMSO (LPS-only treated group).
11S,17S-DiHDHA inhibits MAPK and NF-κB signaling pathway in LPS-stimulated SIM-A9 cells
To investigate the effects of 11S,17S-DiHDHA on mitogen-activated protein kinases (MAPKs) and nuclear factor kappa B (NF-κB) signaling pathways in the suppression of neuroinflammation in SIM-A9 cells, we performed Western blot analysis to examine whether 11S,17S-DiHDHA inhibits LPS-induced activation of ERK1/2, p38, JNK, and p65. As shown in Fig. 4A, LPS treatment alone significantly increased the phosphorylation of ERK1/2, p38, and JNK (key components of the MAPK signaling pathway) compared to the untreated group (CON). However, 11S,17S-DiHDHA markedly suppressed LPS-induced phosphorylation of p38 and JNK in a statistically significant manner. However, 11S,17S-DiHDHA did not exhibit inhibitory effects on LPS-induced ERK1/2 phosphorylation. These findings indicate that 11S,17S-DiHDHA selectively inhibits the activation of p38 and JNK among the key components of the MAPK signaling pathway in neuroinflammation. Furthermore, 11S,17S-DiHDHA suppressed LPS-induced p65 phosphorylation (Fig. 4B), indicating that it inhibits NF-κB activation in neuroinflammation.
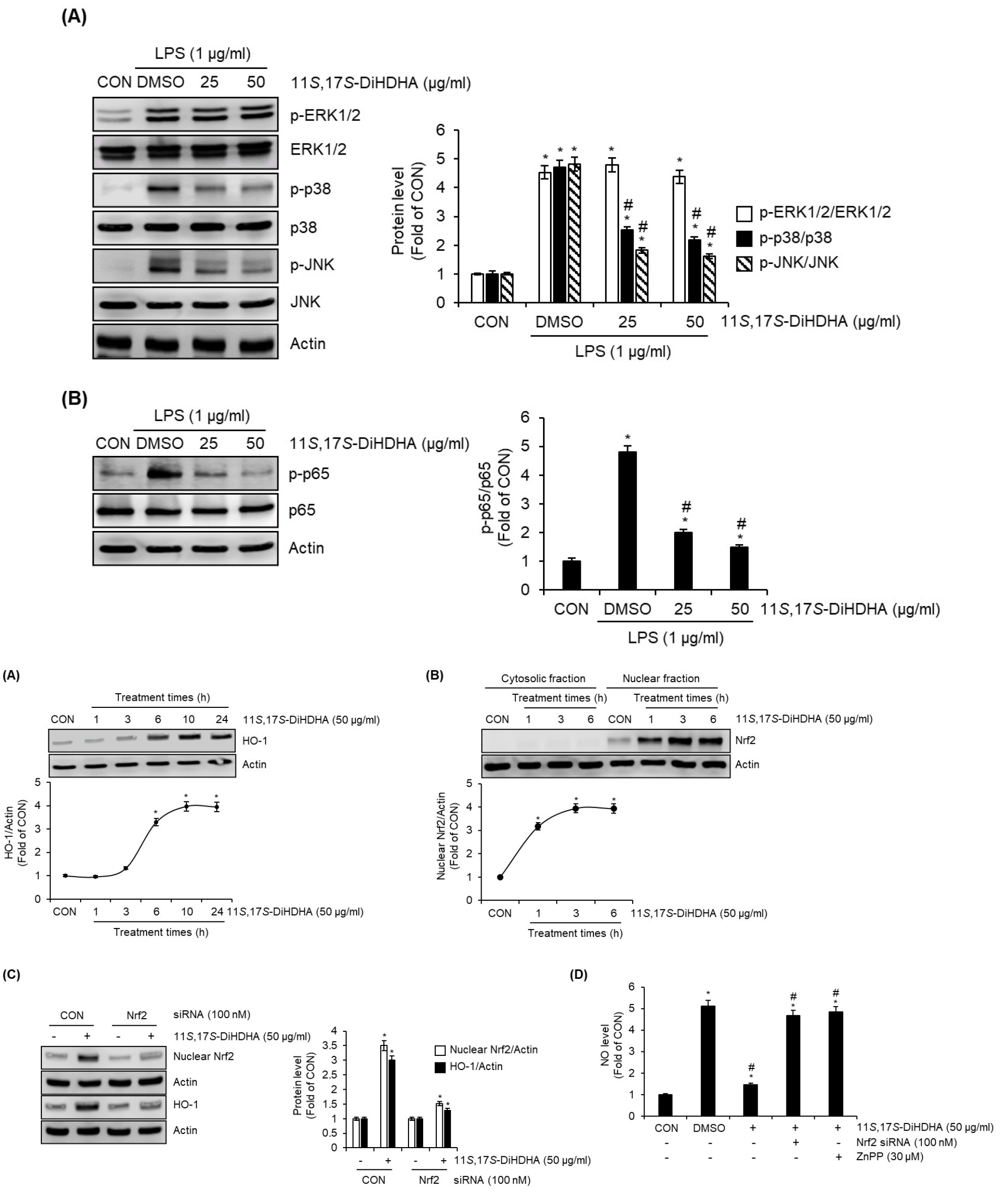
Fig. 4.
Effect of 11S,17S-DiHDHA on MAPK and NF-κB signaling pathways in LPS-stimulated SIM-A9 cells. (A) and (B) SIM-A9 cells were pre-treated with 11S,17S-DiHDHA for 2 h and then co-treated with LPS (1 ㎍/mL) for 30 min. The protein levels such as p-ERK1/2, ERK1/2, p-p38, p38, p-JNK, JNK, p-p65, and p65 were measured using Western blot analysis. *P < 0.05 vs CON (untreated group). #P < 0.05 vs DMSO (LPS-only treated group).
11S,17S-DiHDHA activates Nrf2/HO-1 signaling pathway in SIM-A9 cells
To investigate whether 11S,17S-DiHDHA activates the Nrf2/HO-1 signaling pathway in SIM-A9 cells, we performed a time-course analysis of Nrf2 and HO-1 protein expression following 11S,17S-DiHDHA treatment. Western blot analysis was employed to assess temporal changes in protein levels, providing insight into the regulatory dynamics of Nrf2/HO-1 activation in response to 11S,17S-DiHDHA stimulation. As shown in Fig. 5A and 5B, the protein level of HO-1 in SIM-A9 cells began to increase significantly at 6 hours following 11S,17S-DiHDHA treatment. In contrast, nuclear Nrf2 levels exhibited a significant increase as early as 1 hour post-treatment, suggesting a temporal sequence in the activation of the Nrf2/HO-1 signaling pathway. To determine whether Nrf2 directly contributes to the 11S,17S-DiHDHA-induced upregulation of HO-1 protein levels, we performed Nrf2 knockdown using Nrf2-specific siRNA prior to 11S,17S-DiHDHA treatment and subsequently analyzed HO-1 protein expression via Western blotting analysis. As a result, Nrf2 knockdown effectively suppressed the 11S,17S-DiHDHA-mediated increase in HO-1 protein levels (Fig. 5C), indicating that Nrf2 plays a critical role in the regulation of HO-1 expression in response to 11S,17S-DiHDHA. To investigate whether 11S,17S-DiHDHA-mediated activation of the Nrf2/HO-1 signaling pathway contributes to the suppression of NO production in LPS-stimulated SIM-A9 cells, we performed Nrf2 knockdown using Nrf2-specific siRNA or inhibited HO-1 with ZnPP prior to treatment with 11S,17S-DiHDHA and LPS. The levels of NO were then assessed using the Griess assay. As shown in Fig. 5D, both Nrf2 knockdown and HO-1 inhibition effectively attenuated the 11S,17S-DiHDHA-mediated suppression of NO production, indicating that the Nrf2/HO-1 signaling pathway plays a crucial role in the regulatory mechanism underlying NO inhibition.
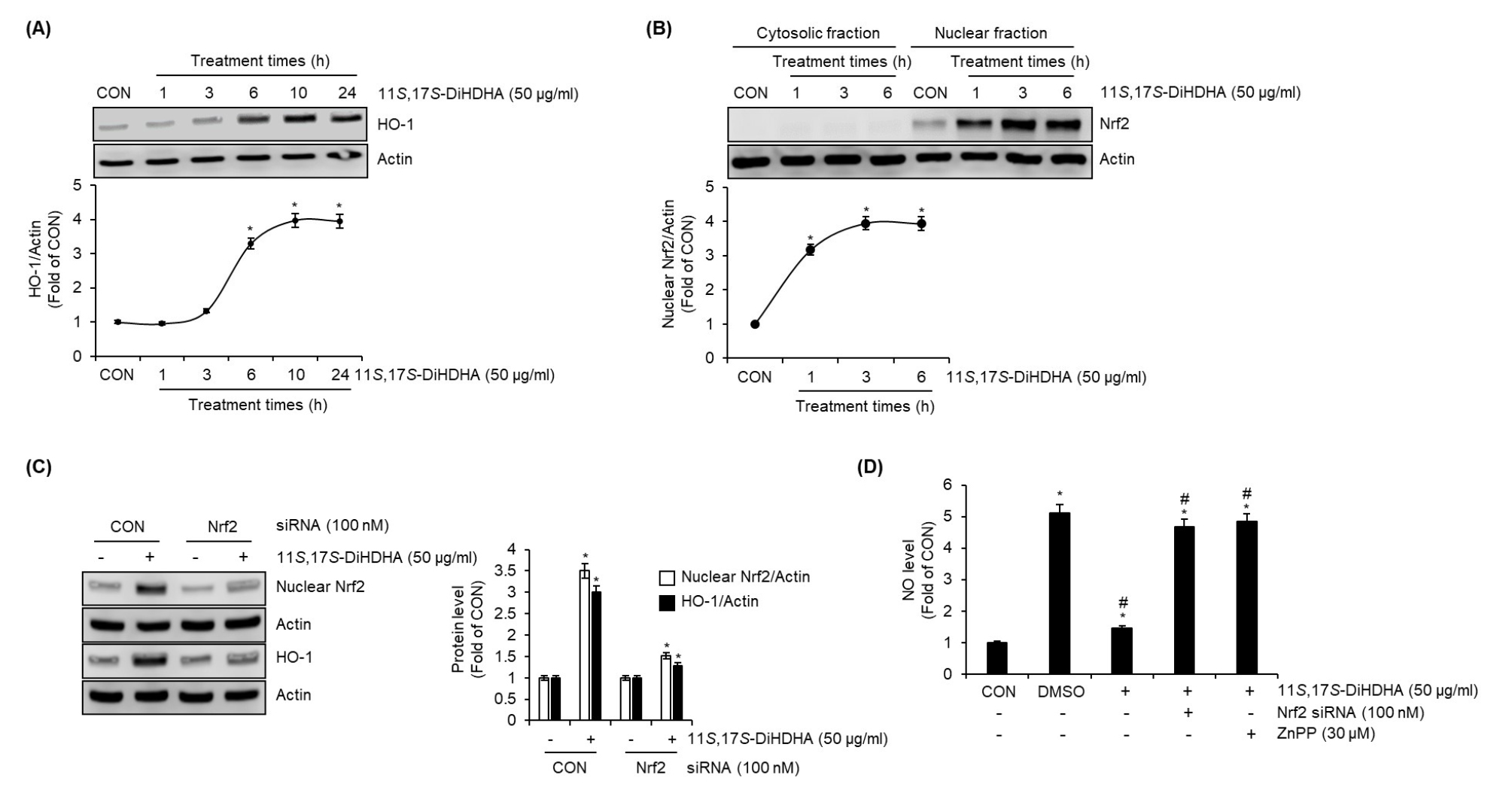
Fig. 5.
Effect of 11S,17S-DiHDHA on the protein level of HO-1 and nuclear Nrf2 in SIM-A9 cells. (A) SIM-A9 cells were treated with 11S,17S-DiHDHA for the indicated times. HO-1 protein level was measured using Western blot analysis. (B) SIM-A9 cells were treated with 11S,17S-DiHDHA for the indicated times. Cytosolic and nuclear fractions were prepared using the Nuclear Extract Kit. Nrf2 protein level was measured using Western blot analysis. (C) SIM-A9 cells transfected with control- or Nrf2-siRNA were treated with 11S,17S-DiHDHA for 1 h for nuclear Nrf2 or 6 h for HO-1, and then total protein for HO-1 and nuclear fraction for Nrf2 were prepared. The protein levels of HO-1 and nuclear Nrf2 were measured using Western blot analysis. (D) SIM-A9 cells transfected with control- or Nrf2-siRNA, or SIM-A9 cells pretreated with ZnPP for 2 h were treated with 11S,17S-DiHDHA for 2 h and then co-treated with LPS for 18 h. NO level was measured using Griess assay. *P < 0.05 vs CON (untreated group). #P < 0.05 vs DMSO (LPS-only treated group).
11S,17S-DiHDHA-mediated activation of Nrf2/HO-1 signaling pathway is dependent on PI3K activation in SIM-A9 cells
To investigate the involvement of PI3K in the activation of the Nrf2/HO-1 signaling pathway mediated by 11S,17S-DiHDHA, we pretreated SIM-A9 cells with LY294002, a selective PI3K inhibitor, prior to 11S,17S-DiHDHA treatment. Western blot analysis revealed that in the absence of PI3K inhibition, 11S,17S-DiHDHA treatment led to a significant increase in nuclear Nrf2 levels, accompanied by an upregulation of HO-1 protein expression. However, this effect was completely abrogated in LY294002-treated cells, indicating that PI3K activity is essential for 11S,17S-DiHDHA-mediated activation of Nrf2/HO-1 signaling pathway (Fig. 6A). Furthermore, to determine whether 11S,17S-DiHDHA modulates PI3K signaling, we assessed the phosphorylation levels of PI3K (p-PI3K), the active form of PI3K. Western blot analysis demonstrated a significant increase in p-PI3K levels following 11S,17S-DiHDHA treatment, suggesting that 11S,17S-DiHDHA activates PI3K signaling (Fig. 6B).
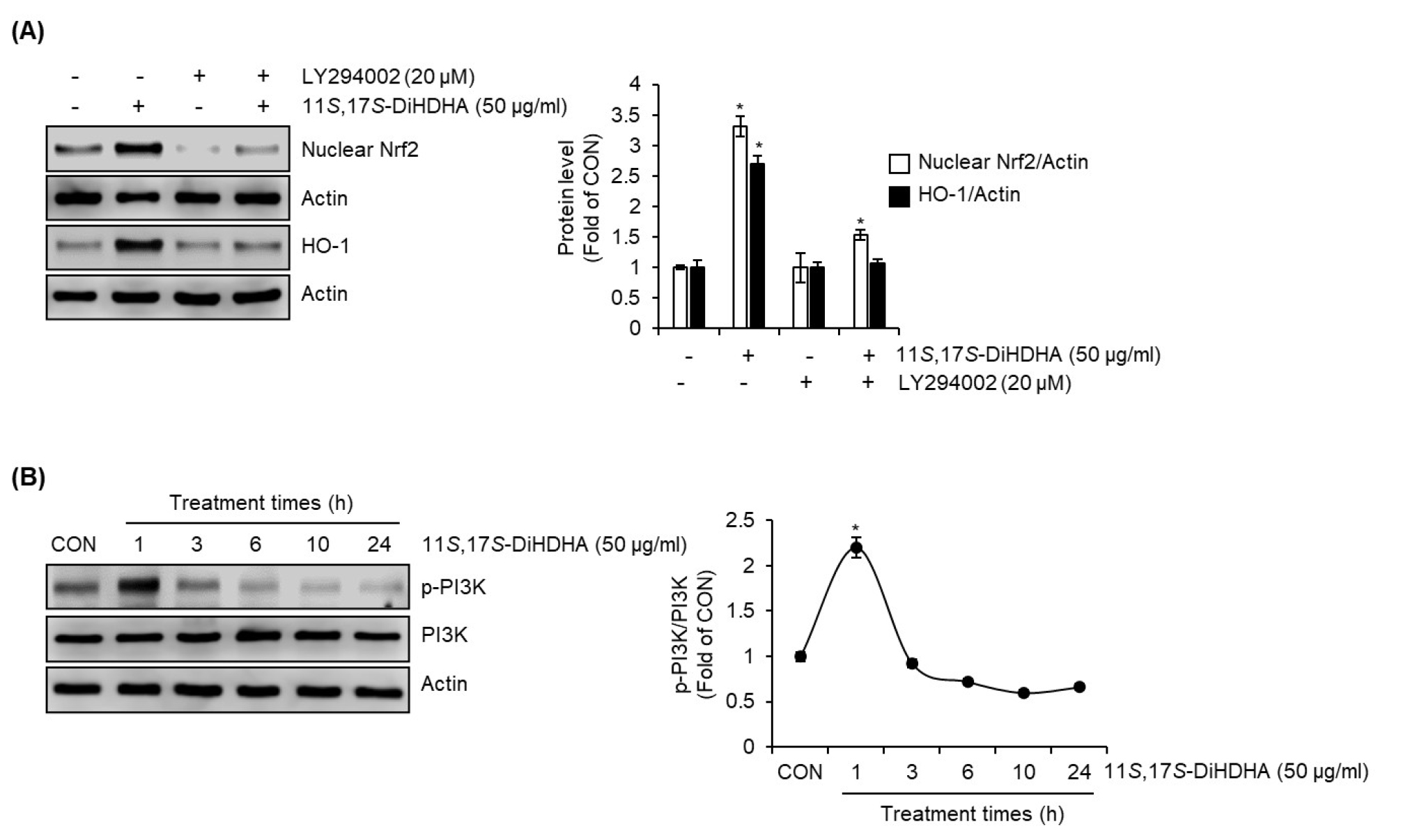
Fig. 6.
Effect of PI3K on 11S,17S-DiHDHA-mediated activation of Nrf2/HO-1 signaling pathway in SIM-A9 cells. (A) SIM-A9 cells were pretreated with LY294002 for 2 h and then co-treated with 11S,17S-DiHDHA for 1 h for Nrf2 or for 6 h for HO-1. Total protein for HO-1 and nuclear fraction for Nrf2 were prepared. The protein levels of HO-1 and nuclear Nrf2 were measured using Western blot analysis. (B) SIM-A9 cells were treated with 11S,17S-DiHDHA for the indicated times. The protein levels of p-PI3K and PI3K were measured using Western blot analysis. *P < 0.05 vs untreated cells or LY294002-only treated cells.
11S,17S-DiHDHA inhibits Aβ25-35-induced cell death in SH-SY5Y cells
To investigate whether 11S,17S-DiHDHA exhibits protective activity against β-amyloid-induced cytotoxicity, we evaluated the extent to which 11S,17S-DiHDHA mitigates Aβ25-35-induced reductions in cell viability in SH-SY5Y cells. As shown in Fig. 7A, treatment with Aβ25-35 alone resulted in a significant reduction in cell viability. However, co-treatment with 11S,17S-DiHDHA markedly attenuated Aβ25-35-induced cytotoxicity, effectively preserving cell survival. Furthermore, Aβ25-35 treatment alone led to a notable increase in lactate dehydrogenase (LDH) release, a key marker of cytotoxicity (Fig. 7B). However, 11S,17S-DiHDHA treatment significantly suppressed this increase (Fig. 7B), indicating its protective effect against Aβ25-35-induced cellular damage. We further investigated the effects of 11S,17S-DiHDHA on the expression levels of apoptosis-related proteins in Aβ25-35-treated cells using Western blot analysis. As shown in Fig. 7C, treatment with Aβ25-35 alone resulted in a significant increase in the pro-apoptotic protein Bax and a decrease in the anti-apoptotic protein Bcl-2 compared to the untreated control. However, 11S,17S-DiHDHA effectively attenuated the Aβ25-35-induced upregulation of Bax and downregulation of Bcl-2, suggesting its potential protective role against apoptosis. Finally, to determine whether the protective effect of 11S,17S-DiHDHA against Aβ25-35-induced SH-SY5Y cell death is mediated through the suppression of ROS, we investigated whether 11S,17S-DiHDHA inhibits Aβ25-35-induced ROS generation. As presented in Fig. 7D, 11S,17S-DiHDHA significantly attenuated the Aβ25-35-induced increase in ROS levels, indicating its potential role in mitigating oxidative stress.
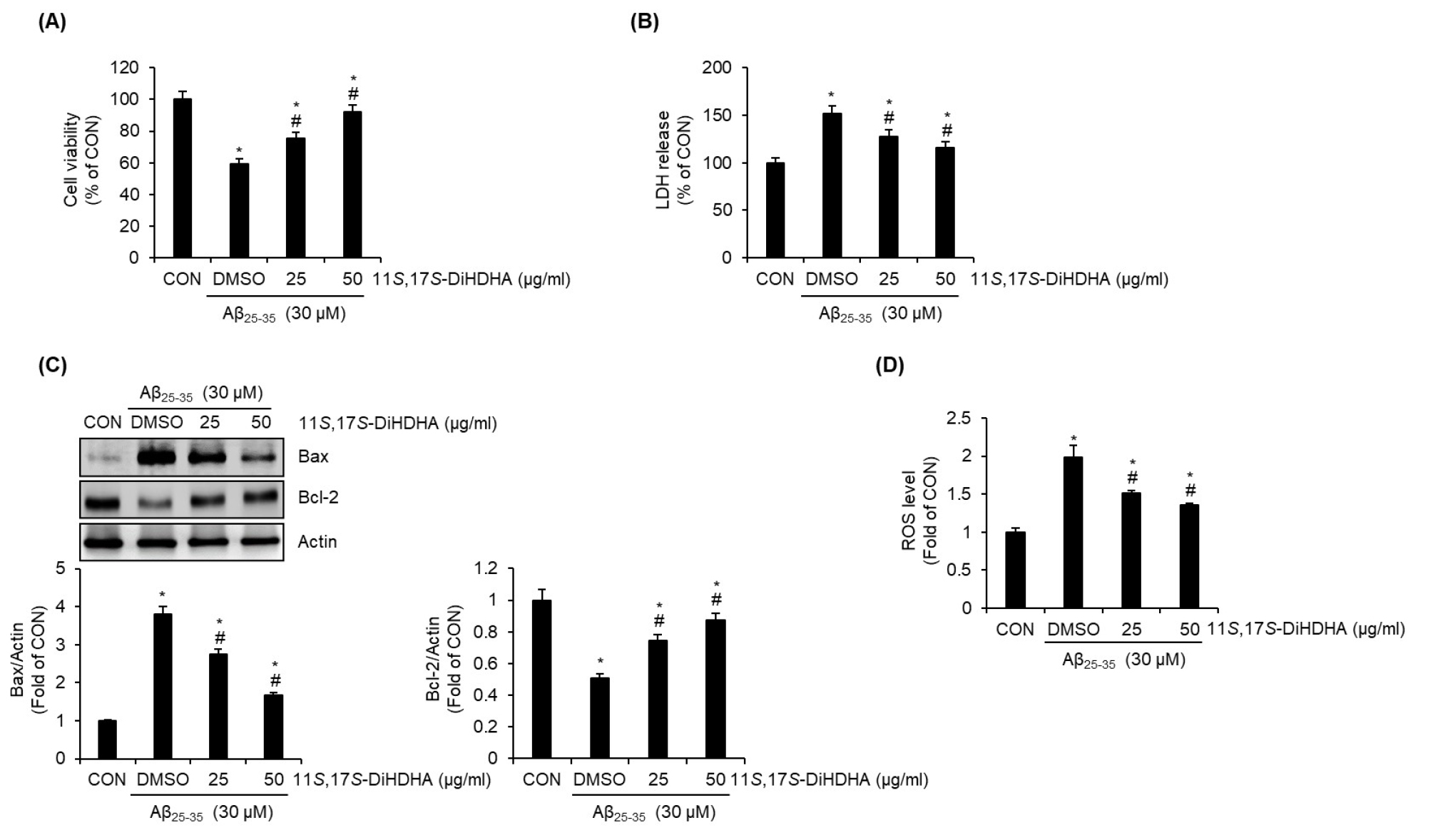
Fig. 7.
Protective effect of 11S,17S-DiHDHA on the Aβ25-35-mediated cell death in SH-SY5Y cells. SH-SY5Y cells were pre-treated with 11S,17S-DiHDHA for 2 h and then co-treated with Aβ25-35 for 24 h. (A) Cell viability was measured using MTT assay. (B) LDH level was measured using the CyQUANT™ LDH Cytotoxicity Assay Kit. (C) The levels of Bax and Bcl-2 protein were measured using Western blot analysis. (D) ROS level was measured using the Intracellular ROS Assay Kit. *P < 0.05 vs CON (untreated group). #P < 0.05 vs DMSO (LPS-only treated group).
Discussion
NO is known to be generated by iNOS during the conversion of L-arginine to L-citrulline (Coleman, 2001; Lee et al., 2025). In the brain, glial cells also produce NO in response to inflammatory stimuli (Olivera et al., 2016). While physiological levels of NO contribute to maintaining brain homeostasis (Liy et al., 2021), excessive NO production has been reported to exacerbate neuroinflammation, leading to neuronal cell death and tissue damage (Olivera et al., 2016). Neurodegenerative diseases, such as AD, remain a significant unresolved health challenge (Liy et al., 2021). Since NO is a key contributor to neurotoxicity, its inhibition is considered a promising therapeutic target for the treatment of neurodegenerative disorders like AD (Liy et al., 2021). In this study, we demonstrated that 11S,17S-DiHDHA suppresses NO overproduction in LPS-stimulated SIM-A9 cells by inhibiting iNOS expression. This finding supports the notion that 11S,17S-DiHDHA possesses anti-neuroinflammatory properties and suggests its potential as a functional candidate for the prevention or treatment of neurodegenerative diseases such as AD.
In this study, we investigated the effects of 11S,17S-DiHDHA on neuroinflammation by evaluating its regulatory role in the MAPK and NF-κB signaling pathways in SIM-A9 cells. Our results demonstrated that 11S,17S-DiHDHA selectively inhibited the LPS-induced phosphorylation of p38 and JNK while having no significant effect on ERK1/2 phosphorylation. In addition, 11S,17S-DiHDHA effectively suppressed the phosphorylation of p65, indicating its inhibitory effect on NF-κB activation. These findings suggest that 11S,17S-DiHDHA exerts its anti-inflammatory effects in microglial cells by modulating key inflammatory signaling pathways, particularly p38, JNK, and NF-κB. The MAPK pathway is a crucial signaling cascade that mediates inflammatory responses in microglial cells (Kaminska et al., 2009). Among its components, p38 and JNK are strongly associated with the production of pro-inflammatory cytokines and neurotoxic mediators in response to LPS stimulation (Kyriakis and Avruch, 2012). The inhibition of these kinases has been shown to alleviate neuroinflammation by suppressing the expression of inflammatory mediators such as TNF-α, IL-6, and iNOS (Chen et al., 2022; Nguyen et al., 2022). Our results align with previous findings that inhibition of p38 and JNK leads to a reduction in microglial activation and inflammatory responses. In contrast, the ERK1/2 pathway has been reported to exhibit dual roles in neuroinflammation, acting as both a pro-inflammatory and neuroprotective signal depending on the cellular context (Cagnol and Chambard, 2010). The observation that 11S,17S-DiHDHA did not inhibit ERK1/2 phosphorylation suggests that its anti-inflammatory action may be primarily mediated through the selective suppression of p38 and JNK rather than global inhibition of MAPK signaling. NF-κB is another pivotal regulator of neuroinflammation, playing a key role in the transcriptional activation of various inflammatory genes (Shih et al., 2015). The phosphorylation of p65, a critical subunit of NF-κB, is required for its nuclear translocation and subsequent induction of inflammatory mediators (Shih et al., 2015). Our findings indicate that 11S,17S-DiHDHA effectively suppressed LPS-induced p65 phosphorylation, suggesting that it inhibits NF-κB activation and thereby attenuates inflammatory gene expression. This result is consistent with previous studies demonstrating that NF-κB inhibition reduces microglial-mediated neuroinflammation and protects against neurodegenerative processes (Chen et al., 2022; Nguyen et al., 2022). The selective inhibition of p38, JNK, and NF-κB by 11S,17S-DiHDHA suggests that it may serve as a promising therapeutic candidate for neuroinflammatory disorders. Given the well-established role of microglial activation in neurodegenerative diseases such as AD, targeting key inflammatory pathways in microglia has been proposed as a potential strategy for disease intervention (Heneka et al., 2015). Taken together, our findings highlight the role of 11S,17S-DiHDHA in selectively modulating p38, JNK, and NF-κB signaling pathways, providing valuable insights into its potential as an anti-neuroinflammatory agent.
In this study, we demonstrated that 11S,17S-DiHDHA activates the Nrf2/HO-1 signaling pathway in SIM-A9 cells and that this activation is dependent on PI3K signaling. Specifically, our results showed that the increase of nuclear Nrf2 level occurred as early as 1 h after 11S,17S-DiHDHA treatment, followed by a significant increase in HO-1 protein expression at 6 h. Knockdown of Nrf2 effectively suppressed 11S,17S-DiHDHA-induced HO-1 expression, confirming that Nrf2 is a key regulator of HO-1 induction in response to 11S,17S-DiHDHA. Furthermore, the inhibition of either Nrf2 or HO-1 abolished the suppressive effect of 11S,17S-DiHDHA on NO production in LPS-stimulated SIM-A9 cells, indicating that the Nrf2/HO-1 pathway plays a crucial role in NO regulation. Additionally, we found that PI3K inhibition with LY294002 prevented 11S,17S-DiHDHA-mediated increase in the level of nuclear Nrf2 and HO-1 protein, demonstrating that PI3K activation is required for the induction of the Nrf2/HO-1 signaling pathway. Finally, we confirmed that 11S,17S-DiHDHA increases PI3K phosphorylation, further supporting the notion that PI3K functions as an upstream regulator of Nrf2/HO-1 activation in response to 11S,17S-DiHDHA. The Nrf2/HO-1 pathway is a well-established defense mechanism against oxidative and inflammatory stress, playing a pivotal role in cellular protection (O’Rourke et al., 2024). Nrf2 activation leads to the transcriptional induction of HO-1, which subsequently exerts cytoprotective effects by attenuating oxidative damage and suppressing inflammatory responses (O’Rourke et al., 2024). The temporal sequence observed in our study, where nuclear Nrf2 accumulation preceded HO-1 upregulation, aligns with previous findings demonstrating that Nrf2 functions as a transcriptional activator of HO-1 (Zhang et al., 2021). Our results further emphasize the importance of Nrf2 in the regulation of HO-1, as Nrf2 knockdown abolished the 11S,17S-DiHDHA-induced increase in HO-1 expression. One of the key findings of this study is the role of the Nrf2/HO-1 pathway in modulating NO production in microglial cells. Excessive NO production is a hallmark of neuroinflammation and has been implicated in the pathogenesis of various neurodegenerative diseases (Olivera et al., 2016). We demonstrated that the inhibition of Nrf2 or HO-1 reversed the NO-suppressive effects of 11S,17S-DiHDHA, providing strong evidence that this pathway plays a regulatory role in NO production. Previous studies have shown that activation of the Nrf2/HO-1 axis leads to a reduction in iNOS expression, thereby limiting NO production and mitigating inflammation (Luo et al., 2018). Our findings support this mechanism and highlight 11S,17S-DiHDHA as a potential therapeutic candidate for neuroinflammatory conditions. Furthermore, our results underscore the essential role of PI3K in 11S,17S-DiHDHA-mediated increase in nuclear Nrf2 and HO-1 protein level. PI3K is known to modulate the activation of Nrf2/HO-1 signaling pathway (Xu et al., 2015). In our study, PI3K inhibition completely abolished 11S,17S-DiHDHA-mediated increase in nuclear Nrf2 and HO-1 protein level, indicating that PI3K is a prerequisite for the activation of Nrf2/HO-1 signaling pathway. Additionally, the observed increase in phosphorylated PI3K levels following 11S,17S-DiHDHA treatment further suggests that PI3K serves as an upstream regulator in this pathway.
In this study, we demonstrated that 11S,17S-DiHDHA exhibits protective effects against β-amyloid (Aβ)-induced cytotoxicity in SH-SY5Y cells by attenuating oxidative stress. Our findings revealed that 11S,17S-DiHDHA effectively mitigated Aβ25-35-induced reductions in cell viability, as evidenced by its ability to preserve cell survival and suppress LDH release. Furthermore, 11S,17S-DiHDHA modulated the expression of apoptosis-related proteins by reducing the pro-apoptotic protein Bax and restoring levels of the anti-apoptotic protein Bcl-2. In addition, we observed that 11S,17S-DiHDHA significantly attenuated Aβ25-35-induced ROS production, suggesting that its cytoprotective effects may be mediated, at least in part, by mitigating oxidative stress. These results provide strong evidence that 11S,17S-DiHDHA plays a neuroprotective role in Aβ-induced toxicity, which is a key pathological feature of AD. Aβ-induced neurotoxicity is widely recognized as a major contributor to the pathogenesis of AD, leading to neuronal apoptosis and oxidative stress-mediated cell damage (Muthaiyah et al., 2011; Selkoe and Hardy, 2016). Thus, mitigating Aβ-induced oxidative stress-mediated neurotoxicity has been recognized as a promising strategy for the prevention of AD (Mairuae et al., 2019). The significant reduction in cell viability and increased LDH release observed in Aβ25-35-treated SH-SY5Y cells in our study aligns with previous reports demonstrating Aβ-induced cytotoxicity in neuronal cells (Mairuae et al., 2019). The ability of 11S,17S-DiHDHA to restore cell viability and suppress LDH release strongly suggests that it confers protection against Aβ-induced cell death. Aβ-induced apoptosis promotes neuronal cell death, which is widely recognized as a key contributing factor in the pathogenesis of AD (Kumari et al., 2023). The balance between pro-apoptotic (Bax) and anti-apoptotic (Bcl-2) proteins is a key determinant of neuronal survival, with an increased Bax/Bcl-2 ratio promoting apoptosis (Reed, 1994). Our results demonstrated that 11S,17S-DiHDHA effectively counteracted Aβ25-35-induced apoptosis by downregulating Bax expression and restoring Bcl-2 levels. Oxidative stress is another critical factor contributing to Aβ-induced neuronal damage. Aβ accumulation triggers excessive ROS production, leading to oxidative damage, mitochondrial dysfunction, and further exacerbation of neuroinflammation (Cheignon et al., 2018). In our study, 11S,17S-DiHDHA significantly suppressed Aβ25-35-induced ROS generation, highlighting its potential antioxidant role in mitigating Aβ-mediated toxicity. Taken together, our study provides compelling evidence that 11S,17S-DiHDHA protects against Aβ-induced cytotoxicity by modulating apoptosis-related proteins and attenuating oxidative stress. These findings suggest that 11S,17S-DiHDHA may serve as a potential preventive candidate for mitigating Aβ-mediated neurotoxicity in AD.
In this study, we demonstrated that 11S,17S-DiHDHA exerts neuroprotective effects through its anti-inflammatory properties and inhibition of neuronal cell death. In LPS-stimulated SIM-A9 microglial cells, 11S,17S-DiHDHA significantly reduced NO production by downregulating iNOS expression through inhibiting MAPK (p38, JNK) and NF-κB signaling pathways and activating the PI3K/Nrf2/HO-1 signaling cascade. In Aβ25–35-treated SH-SY5Y neuronal cells, 11S,17S-DiHDHA effectively alleviated neuronal damage by preserving cell viability, reducing LDH release, and regulating the expression of apoptosis-related proteins. Taken together, these findings highlight the anti-neuroinflammatory and neuroprotective properties of 11S,17S-DiHDHA and suggest that this previously uncharacterized 11S,17S-DiHDHA may interrupt key pathological mechanisms of Alzheimer’s disease. Although this study presents promising findings, a key limitation is that it was conducted using in vitro models. Data obtained from in vitro experiments may not fully recapitulate the complex mechanisms of neuroinflammation and neurodegeneration in a physiological environment. Therefore, further validation in in vivo models of neuroinflammation and AD is essential to assess the therapeutic efficacy and bioavailability of 11S,17S-DiHDHA.
