

Introduction
Materials and Methods
Chemical reagents
Sample preparation
Quantification of major immunostimulatory phytochemical constituents
Cell culture
MTT assay
Griess assay for NO level
Measurement of IL-1β, IL-6 and TNF-α
Neutral Red assay for phagocytotic activity
Measurement of ROS level
Reverse transcription polymerase chain reaction (RT- PCR)
SDS-PAGE and western blot analysis
Statistical analysis
Results
SRF induces macrophage activation in RAW264.7 cells
TLR4 is essential for SRF-induced macrophage activation in RAW264.7 cells
SRF induces macrophage activation via TLR4-dependent activation of JNK and NF-κB signaling pathways in RAW264.7 cells
SRF induces ROS production through TLR4, contributing to JNK and NF-κB activation and subsequent NO production in RAW264.7 cells
Quantification of major immunostimulatory phytochemical constituents in SRF
Discussion
Introduction
Innate immunity serves as the first line of defense against invading pathogens and plays a crucial role in maintaining immune homeostasis (Wang et al., 2024a). Among the various innate immune cells, macrophages act as central effectors that recognize, engulf, and eliminate microorganisms and cellular debris through phagocytosis (Marshall et al., 2018). In addition to their antimicrobial activity, macrophages secrete pro- inflammatory mediators, including nitric oxide (NO), reactive oxygen species (ROS), and cytokines, which orchestrate broader immune responses (Marshall et al., 2018; Herb and Schramm, 2021). Enhancing macrophage activity is therefore considered a potential strategy to improve host defense mechanisms (Chen et al., 2023).
Macrophage activation is tightly regulated by pattern recognition receptors such as Toll-like receptor 4 (TLR4), which detects pathogen-associated molecular patterns (PAMPs) like lipopolysaccharide (LPS) (Kim et al., 2023). Upon stimulation, TLR4 triggers intracellular ROS generation and activates downstream signaling cascades including the mitogen-activated protein kinase (MAPK) pathways and nuclear factor kappa B (NF-κB) (Deng et al., 2015). These pathways are known to induce the expression of inducible nitric oxide synthase (iNOS), thereby promoting NO production and enhancing antimicrobial and phagocytic functions (Deng et al., 2015). Thus, targeting these signaling mechanisms is a promising approach for evaluating the immunostimulatory potential of natural substances (Tabarsa et al., 2020).
Natural products have long served as a valuable source of therapeutic agents due to their diverse bioactivities and relatively low toxicity (Ahmad et al., 2025). In recent years, interest has grown in discovering plant-derived compounds that can modulate innate immune responses (Sotto et al., 2020). Several plant extracts and phytochemicals have demonstrated the ability to activate macrophages (Cui et al., 2020; Tabarsa et al., 2022; Yang et al., 2022). However, the repertoire of known immunostimulatory natural substances remains limited. Continued exploration of underutilized medicinal plants is critical for identifying new candidates that can promote immune function.
Sambucus racemosa subsp. pendula (SR) commonly referred to as Ulleungdo elder or Korean elder, is a member of the Caprifoliaceae family and the genus Sambucus. This taxon is considered endemic to Korea and is reportedly restricted in its distribution to Ulleungdo Island (Lim, 2022). Recently, we demonstrated that the aqueous extract of SR leaves exerts immunostimulatory activity and anti-obesity activity (Choi et al., 2024; Choi et al., 2025). The berries of Sambucus, commonly known as elderberries, are well-recognized for their health-promoting potential, attributed to their antioxidant, antiviral, anticancer, and immunostimulatory activities (Onolbaatar et al., 2025). However, to date, research on the health-promoting properties of elderberries has been largely limited to Sambucus nigra L., commonly known as black elderberry (Liu et al., 2022). In contrast, studies on the fruit of SR, commonly known as red elderberry, remain virtually nonexistent.
In this study, we investigated the macrophage-activating properties of the fruits of Sambucus racemosa subsp. pendula (SRF) using the RAW264.7 murine macrophage cell line. We focused on elucidating the molecular mechanism underlying its immunostimulatory effect, particularly the involvement of the TLR4/ROS/JNK and NF-κB/iNOS/NO axis in the enhancement of phagocytic activity. Our findings provide mechanistic insights into the immunostimulatory effects of SRF and support its potential as a functional natural product for innate immune enhancement.
Materials and Methods
Chemical reagents
The reagents (3-(4,5-Dimethylthiazol-2-yl)-2, 5-Diphenyltetrazolium Bromide) (MTT; cat. no. 475989), Neutral red (cat. no. N4638), and Griess reagent (cat. no. G4410) were purchased from Sigma-Aldrich (St. Louis, Missouri, USA). Chemical inhibitors such as PD98059 (cat. no. 513000), SB203580 (cat. no. 58307), SP600125 (cat. no. 55567), BAY 11-7082 (cat. no. 196870), N-acetyl-l-cysteine (NAC; cat. no. A9165), and TAK-242 (cat. no. 614316) were purchased from Sigma-Aldrich (St. Louis, MO, USA). C29 (cat. no. 27029) was purchased from Cayman Chemical Company (Ann Arbor, MI, USA). The primary antibodies such as c-Jun N-terminal kinases (JNK, cat. no: 9258), p-JNK (cat. no: 9251), p65 (cat. no: 8242), p-p65 (cat. no. 3033), and β-actin (cat. no: 5125) were purchased from Cell Signaling Technology (Danvers, MA, USA). The secondary antibody for anti- rabbit IgG, HRP-linked antibody (cat. no: 7074) was purchased from Cell Signaling Technology (Danvers, MA, USA).
Sample preparation
Fruits of Sambucus racemosa subsp. pendula (Voucher number: FMRC-250526-03-01-5323~5326) were collected from Ulleung Island, Korea, in May 2025. Botanical identification of the plant material was conducted by the National Institute of Forest Science, Korea. Fruits of Sambucus racemosa subsp. pendula were washed thoroughly with distilled water, freeze-dried, and subsequently pulverized into a fine powder. For extraction, 5 g of the powdered fruits of Sambucus racemosa subsp. pendula was mixed with 100 mL of distilled water and subjected to shaking at 150 rpm for 24 h at 20°C. The resulting mixture was centrifuged at 15,000 rpm to remove insoluble components, and the clear supernatant was carefully collected. This aqueous extract was then lyophilized to yield the final test sample, designated as SRF. The SRF powder was stored at -80°C until use and was reconstituted in distilled water immediately before treatment of cells.
Quantification of major immunostimulatory phytochemical constituents
The total polysaccharide content of SRF was determined using the phenol-sulfuric acid colorimetric method with slight modifications. Briefly, an aliquot of the sample solution (200 μL) was mixed with 1 mL of 5% (w/v) phenol solution, followed by the addition of 5 mL of concentrated sulfuric acid. The reaction mixture was allowed to stand at room temperature for 20 min to allow full color development. The absorbance was then measured at 490 ㎚ using a microplate reader (SpectraMax M2, Molecular Devices, San Jose, CA, USA). A standard calibration curve was constructed using D-(+)-mannose and the total polysaccharide content was expressed as milligrams of mannose equivalent per gram of extract (㎎ ME/g extract). The total polyphenol content of SRF was analyzed using the Folin-Ciocalteu colorimetric assay with minor modifications. In brief, 10 μL of the sample solution was mixed with 10 μL of 1 N Folin-Ciocalteu reagent and incubated for 5 min at room temperature. Then, 200 μL of 2% sodium carbonate (Na2CO3) solution was added, and the mixture was further incubated for 30 min in the dark. The absorbance was measured at 760 ㎚ using a microplate reader (SpectraMax M2, Molecular Devices, San Jose, CA, USA). Gallic acid was used to generate a standard calibration curve, and the results were expressed as milligrams of tannic acid equivalent per gram of extract (㎎ TAE/g extract). The total flavonoid content of SRF was measured using an aluminum chloride colorimetric assay with slight modifications. Briefly, 100 μL of the sample solution was combined with 6 μL of 5% sodium nitrite (NaNO2) solution. After a 5 min incubation at room temperature, 12 μL of 10% aluminum chloride (AlCl3) was added, and the mixture was allowed to react for another 6 min. Then, 40 μL of 1 M sodium hydroxide (NaOH) was added, and the mixture was allowed to react for another 11 min. The absorbance was recorded at 510 ㎚ using a microplate reader. A standard curve was established using Rutin, and the results were expressed as milligrams of quercetin equivalent per gram of extract (㎎ RE/g extract).
Cell culture
The murine macrophage cell line RAW264.7 (cat. no. TIB- 71) was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM)/F-12 supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 1% penicillin-streptomycin. Cultures were maintained at 37°C in a humidified incubator with 5% CO2. Subculturing was performed when the cells reached approximately 80% confluence. All experiments were conducted with cells at passage numbers below 15 to ensure cellular integrity and reproducibility.
MTT assay
To assess the effect of SRF on the viability of RAW264.7 cells, a colorimetric assay using MTT was conducted. RAW264.7 cells were seeded into 96-well plates at a density of 1 × 105 cells/well and allowed to adhere overnight. After treatment with SRF for 24 h, 10 μL of MTT solution (final concentration: 0.5 ㎎/mL) was added to each well, followed by incubation for 3 h at 37°C under dark conditions. Subsequently, the supernatant was carefully removed, and 100 μL of DMSO was added to solubilize the resulting formazan crystals. The absorbance was measured at 570 ㎚ using an UV-visible spectrophotometer (SpectraMax M2, Molecular Devices, San Jose, CA, USA). Cell viability was expressed as a percentage relative to the untreated control group (CON).
Griess assay for NO level
The production of NO in RAW264.7 cells was indirectly measured by detecting nitrite levels in the culture medium using the Griess reaction. RAW264.7 cells (1 × 10⁵ cells per well) were seeded into 96-well plates and incubated at 37°C for 24 h. Following this, the cells were treated with SRF and incubated for an additional 24 h at 37°C. For inhibitor experiments, RAW264.7 cells were pre-treated with C29 (100 μM), TAK-242 (10 μM), PD98059 (20 μM), SB203580 (20 μM), SP600125 (20 μM), BAY 11-7082 (20 μM) or NAC (10 mM) for 2 h at 37°C, followed by co-treatment with the SRF for 24 h at the same temperature. Following the treatment period, 100 μL of the supernatant from each well was transferred to a new 96-well plate. An equal volume (100 μL) of freshly prepared Griess reagent was added to each sample. The mixture was gently agitated and incubated for 15 min at room temperature in the dark. Absorbance was then measured at 540 ㎚ using an UV-visible spectrophotometer (SpectraMax M2, Molecular Devices, San Jose, CA, USA). NO level was expressed as a fold induction to the untreated control group (CON).
Measurement of IL-1β, IL-6 and TNF-α
RAW264.7 cells (1 × 10⁵ cells per well) were seeded into 96-well plates and incubated at 37°C for 24 h. Following this, the cells were treated with SRF and incubated for an additional 24 h at 37°C. After SRF treatment, the contents of IL-1β, IL-6, and TNF-α were measured using a Mouse IL-1 beta ELISA Kit (cat. no. BMS6002, Invitrogen, Waltham, MA, USA), Mouse IL-6 ELISA Kit (cat. no. KMC0061, Invitrogen, Waltham, MA, USA), and Mouse TNF alpha ELISA Kit (BMS607-3, Invitrogen, Waltham, MA, USA), according to the manufacturer’s protocol.
Neutral Red assay for phagocytotic activity
Phagocytic activity of RAW264.7 macrophages was evaluated by the Neutral Red uptake method. Briefly, RAW264.7 cells (1 × 10⁵ cells/well) were plated in 96-well culture plates and allowed to adhere for 24 h at 37°C. The cells were then treated with SRF for an additional 24 h under the same conditions. For inhibition assays, cells were pre-incubated with either C29 (100 μM) or TAK-242 (10 μM) for 2 h at 37°C before being co-treated with SRF for 24 h. Following the treatment period, 0.01% Neutral Red solution was added to each well and incubated for 2 h at 37°C. After staining, unincorporated dye was removed, and the dye retained within the cells was solubilized using a lysis buffer composed of 50% ethanol and 1% acetic acid. The absorbance of the resulting solution was measured at 540 ㎚ using a UV-visible spectrophotometer (SpectraMax M2, Molecular Devices, San Jose, CA, USA) to quantify phagocytic activity.
Measurement of ROS level
RAW264.7 cells (1 × 10⁵ cells/well) were plated in 96-well culture plates and allowed to adhere for 24 h at 37°C. The cells were pretreated with the TLR4 inhibitor TAK-242 (10 μM) for 2 h and then co-treated with SRF (100 ㎍/mL) for 24 h. After SRF treatment, ROS level as measured using an OxiSelectTM Intracellular ROS Assay Kit (Green Fluorescence) (cat. no. STA-342, Cell Biolabs, Inc., San Diego, CA, USA) according to the manufacturer’s protocol.
Reverse transcription polymerase chain reaction (RT- PCR)
Total RNA was isolated from RAW264.7 cells using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The concentration and purity of the extracted RNA were determined using a GeneQuant 1300 spectrophotometer (Biochrom, MA, USA) by measuring absorbance at 260 and 280 nm. One microgram of RNA was reverse transcribed into complementary DNA (cDNA) using the Verso cDNA Kit (Thermo Fisher Scientific, Waltham, MA, USA). The resulting cDNA was used as a template for PCR amplification using the PCR Master Mix Kit (Promega, Madison, WI, USA) and iNOS or GAPDH- specific primers. The primer sequences used for iNOS and GAPDH in this study were as follows: iNOS mRNA, F: 5′- GTTACCATGAGGCTGAAATCC-3′ and R: 5′-CCTCTT GTCTTTGACCCAGTAC-3′, GAPDH mRNA, F: 5′-GGAC CTCATGGCCTACATGG-3′ and R: 5′- TAGGGCCTCTCT TGCTCAGT-3′. PCR reactions were carried out using a thermal cycler under the following conditions: initial denaturation at 95°C for 5 minutes, followed by 30 cycles of denaturation at 95°C for 30 seconds, annealing at optimized temperatures for 30 seconds, and extension at 72°C for 30 seconds. PCR products were separated by agarose gel electrophoresis and visualized using a gel documentation system. The intensity of the visualized mRNA bands was quantified using UN-SCAN-IT gel software version 5.1 (Silk Scientific Inc., Vineyard, UT, USA).
SDS-PAGE and western blot analysis
Proteins were extracted from RAW264.7 cells using RIPA lysis buffer (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with protease and phosphatase inhibitors. Following quantification using a Pierce™ BCA Protein Assay Kits (Thermo Fisher Scientific, Waltham, MA, USA), equal amounts of protein (30 ㎍/well) from each sample were mixed with loading buffer and denatured by heating at 95°C for 10 min. Proteins were then separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and subsequently transferred onto a nitrocellulose membrane (Thermo Fisher Scientific, Waltham, MA, USA). The membranes were blocked with 5% non-fat dry milk in TBST (Tris-buffered saline containing 0.1% Tween-20) for 1 h at room temperature to prevent nonspecific binding. After blocking, the membranes were incubated overnight at 4°C with primary antibodies targeting specific proteins, such as JNK, p-JNK, p65, and p-p65. After washing, the membranes were incubated with appropriate HRP-conjugated secondary antibodies for 1 h at room temperature. Immunoreactive bands were visualized using an ECL Select Western Blotting Detection Reagent (cat. no. RPN2232; Cytiva, Wilmington, DE, USA) and detected with an LI-COR C-DiGit Blot Scanner (LI-COR Biosciences, Lincoln, NE, USA). The protein band intensities were quantified using the UN- SCAN-IT gel analysis software, version 5.1 (Silk Scientific Inc., Vineyard, UT, USA).
Statistical analysis
All experiments were repeated at least three times. Statistical analysis was performed using GraphPad Prism version 5.0 (GraphPad Software, Inc.) and data are presented as the mean ± standard deviation. Data was analyzed using one‑way analysis of variance followed by Bonferroni's post hoc test. P<0.05 was considered to indicate a statistically significant difference.
Results
SRF induces macrophage activation in RAW264.7 cells
To evaluate the immunostimulatory potential of SRF, we investigated its ability to activate RAW264.7 macrophages. Treatment of the cells with various concentrations of SRF resulted in a dose-dependent increase in NO production, as determined by the Griess assay (Fig. 1A). Consistently, RT-PCR analysis revealed that SRF significantly upregulated the expression of iNOS in a dose-dependent manner (Fig. 1B), suggesting that SRF-mediated NO production is associated with enhanced iNOS transcription. In addition to NO, the secretion of key pro-inflammatory cytokines (IL-1β, IL-6, and TNF-α) was also markedly increased in response to SRF treatment in a dose-dependent fashion (Fig. 1C). These findings demonstrate that SRF can stimulate macrophage- mediated immune responses by promoting the expression of major cytokines involved in immune signaling. Furthermore, the phagocytic activity of RAW264.7 cells was significantly enhanced following SRF treatment (Fig. 1D), indicating that SRF not only modulates immunostimulatory factors but also augments macrophage functional activity. Importantly, a cell viability assay showed that SRF did not affect the survival of macrophages at the tested concentrations (Fig. 1E). Together, these results suggest that SRF activates macrophages by increasing the secretion of immunostimulatory factors (NO, iNOS, IL-1β, IL-6, and TNF-α) and phagocytic function without compromising cell viability.
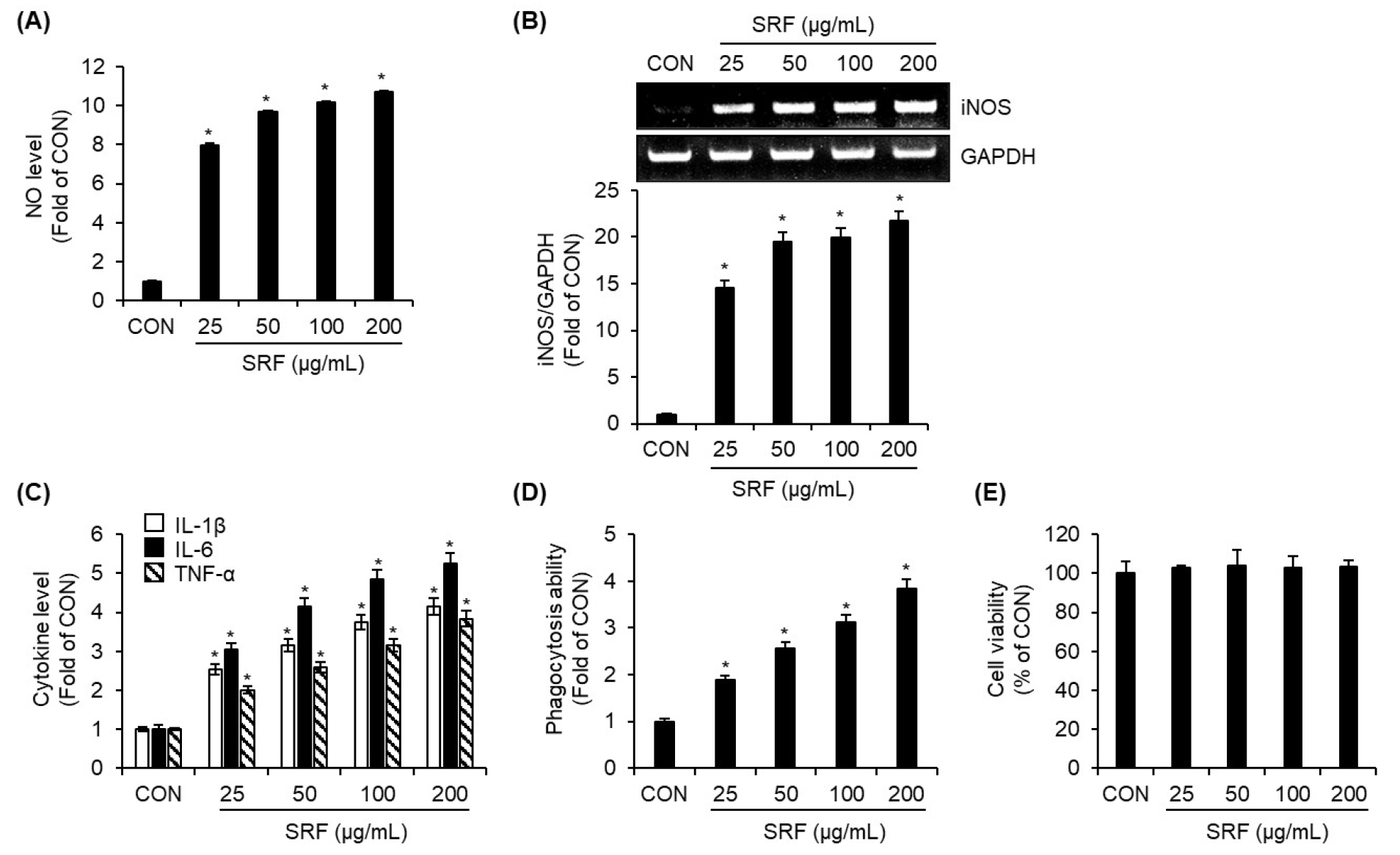
Fig. 1.
Immunostimulatory effects of SRF on RAW264.7 cells. RAW264.7 cells were treated with SRF at the indicated concentrations for 24 h. (A) NO level was quantified by the Griess assay. (B) mRNA expression of iNOS was determined by RT-PCR. (D) Levels of (IL-1β, IL-6, and TNF-α were measured by ELISA Kit. (D) Phagocytic activity was assessed using the Neutral Red uptake assay. (E) Cell viability was evaluated by the MTT assay. *P<0.05 vs. CON (untreated group).
TLR4 is essential for SRF-induced macrophage activation in RAW264.7 cells
To identify the pattern recognition receptor responsible for SRF-mediated macrophage activation, we investigated the roles of TLR2 and TLR4 using their respective inhibitors- C29 (TLR2 inhibitor) and TAK-242 (TLR4 inhibitor). RAW264.7 cells were pretreated with each inhibitor prior to SRF treatment, and the resulting changes in NO production, iNOS expression, and phagocytic activity were analyzed. Inhibition of TLR2 by C29 did not affect SRF-induced NO production (Fig. 2A), iNOS mRNA expression (Fig. 2B), or enhancement of phagocytosis (Fig. 2C), indicating that TLR2 is not involved in mediating the immunostimulatory effects of SRF. In contrast, pretreatment with the TLR4 inhibitor TAK-242 completely abrogated the SRF-induced increase in NO production (Fig. 2A) and iNOS expression (Fig. 2B). Furthermore, TAK-242 significantly suppressed the SRF-mediated enhancement of phagocytic activity (Fig. 2C). These results suggest that TLR4, but not TLR2, is required for SRF-induced macrophage activation. This indicates that SRF exerts its immunostimulatory effects through a TLR4-dependent signaling pathway.
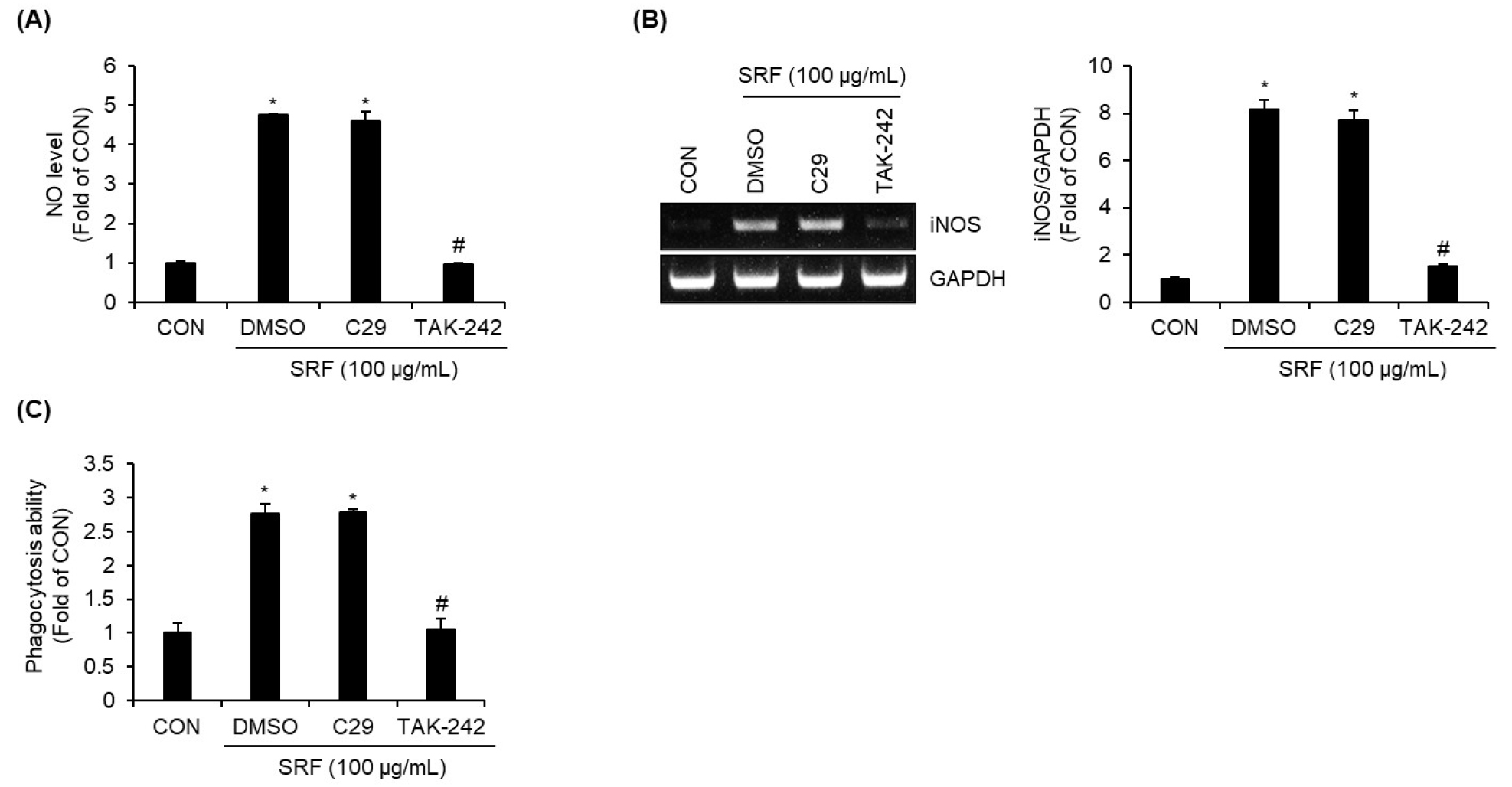
Fig. 2.
TLR4 is essential for SRF-induced macrophage activation in RAW264.7 cells. RAW264.7 macrophages were pretreated with the TLR2 inhibitor C29 (100 μM) or the TLR4 inhibitor TAK-242 (10 μM) for 2 h and then co-treated with SRF (100 ㎍/mL) for 24 h. (A) NO level was measured using the Griess assay. (B) mRNA expression of iNOS was analyzed by RT-PCR. (C) Phagocytic activity was evaluated using the Neutral Red uptake assay. *P<0.05 vs. CON (untreated group), #P<0.05 vs DMSO (SRF-treated group).
SRF induces macrophage activation via TLR4-dependent activation of JNK and NF-κB signaling pathways in RAW264.7 cells
To elucidate the intracellular signaling pathways involved in SRF-mediated macrophage activation downstream of TLR4, we examined the effects of specific signaling inhibitors on NO production and iNOS expression. RAW264.7 cells were pretreated with MAPK pathway inhibitors-PD98059 (ERK 1/2 inhibitor), SB203580 (p38 inhibitor), SP600125 (JNK inhibitor)-as well as BAY 11-7082 (NF-κB inhibitor) prior to SRF stimulation. As shown in Fig. 3A, the inhibition of ERK1/2 with PD98059 or p38 with SB203580 had no significant effect on SRF-induced NO production. In contrast, the inhibition of JNK with SP600125 and inhibition of NF-κB with BAY 11-7082 significantly suppressed the increase in NO production triggered by SRF. Furthermore, the inhibition of both JNK and NF-κB significantly reduced the SRF-induced upregulation of iNOS expression (Fig. 3B), indicating that the JNK and NF-κB pathways play critical roles in SRF-mediated macrophage activation. To confirm the activation of these pathways, the phosphorylation status of JNK (p-JNK) and the NF-κB p65 subunit (p-p65) was assessed over time following SRF treatment. As shown in Fig. 3C, phosphorylation of both JNK and p65 increased from 1 h after SRF stimulation, suggesting the activation of these signaling molecules. Moreover, pretreatment with the TLR4 inhibitor TAK-242 attenuated SRF-induced phosphorylation of both p-JNK and p-p65 (Fig. 3D), confirming that SRF activates the JNK and NF-κB pathways in a TLR4-dependent manner. These results collectively demonstrate that SRF induces macrophage activation through the activation of TLR4-mediated JNK and NF-κB signaling pathways.
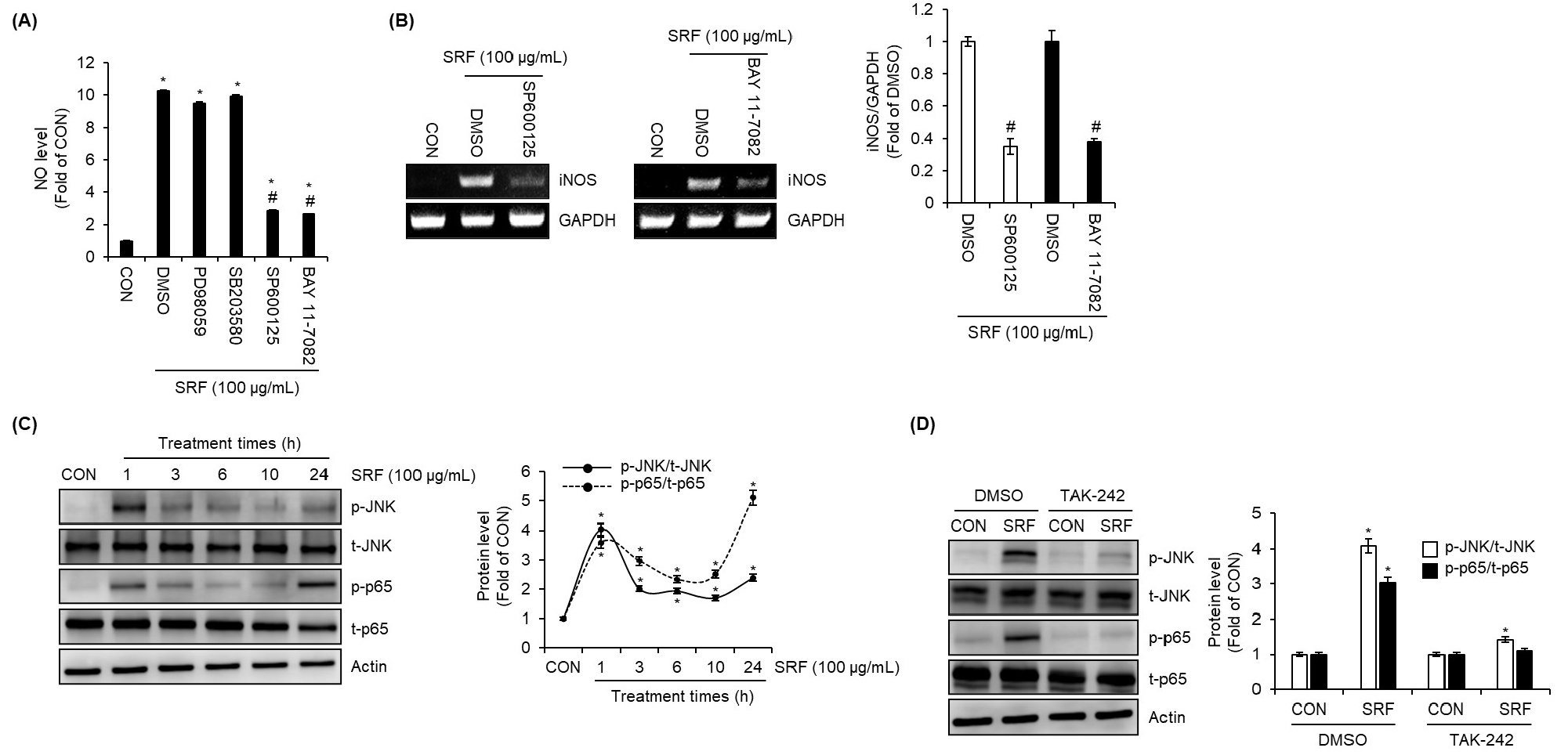
Fig. 3.
SRF activates macrophages via TLR4-dependent JNK and NF-κB signaling pathways in RAW264.7 cells. RAW264.7 cells were pretreated with PD98059 (20 μM; ERK1/2 inhibitor), SB203580 (20 μM; p38 inhibitor), SP600125 (20 μM; JNK inhibitor), or BAY 11-7082 (20 μM; NF-κB inhibitor) for 2 h and then co-treated with SRF (100 ㎍/mL) for 24 h. (A) NO level was measured by the Griess assay, and (B) mRNA expression of iNOS was determined by RT-PCR. (C) RAW264.7 cells were stimulated with SRF (100 ㎍/mL) for the indicated times, and phosphorylation of JNK (p-JNK) and p65 (p-p65) was analyzed by Western blot analysis. (D) RAW264.7 cells were pretreated with the TLR4 inhibitor TAK-242 (10 μM) for 2 h and then co-treated with SRF (100 ㎍/mL) for 1 h, and p-JNK and p-p65 were assessed by Western blot analysis. *P<0.05 vs. CON (untreated group), #P<0.05 vs DMSO (SRF-treated group).
SRF induces ROS production through TLR4, contributing to JNK and NF-κB activation and subsequent NO production in RAW264.7 cells
To investigate whether ROS mediate SRF-induced activation of the JNK and NF-κB signaling pathways in macrophages, we pretreated RAW264.7 cells with the ROS scavenger NAC prior to SRF stimulation. In the absence of NAC, SRF increased the phosphorylation levels of both JNK and the NF-κB p65 subunit. However, NAC pretreatment effectively suppressed SRF-induced phosphorylation of both signaling proteins (Fig. 4A), indicating that ROS production is required for SRF-mediated activation of these pathways. We next examined whether SRF directly promotes ROS generation. As shown in Fig. 4B, SRF significantly increased intracellular ROS levels. Notably, pretreatment with the TLR4 inhibitor TAK-242 reduced SRF-induced ROS generation, suggesting that TLR4 mediates ROS production in response to SRF. Furthermore, the inhibition of ROS production by NAC markedly decreased NO production in SRF-treated cells (Fig. 4C). These findings demonstrate that ROS play a critical intermediary role in the SRF–TLR4 signaling axis, facilitating downstream activation of JNK and NF-κB and ultimately contributing to NO production in macrophages.
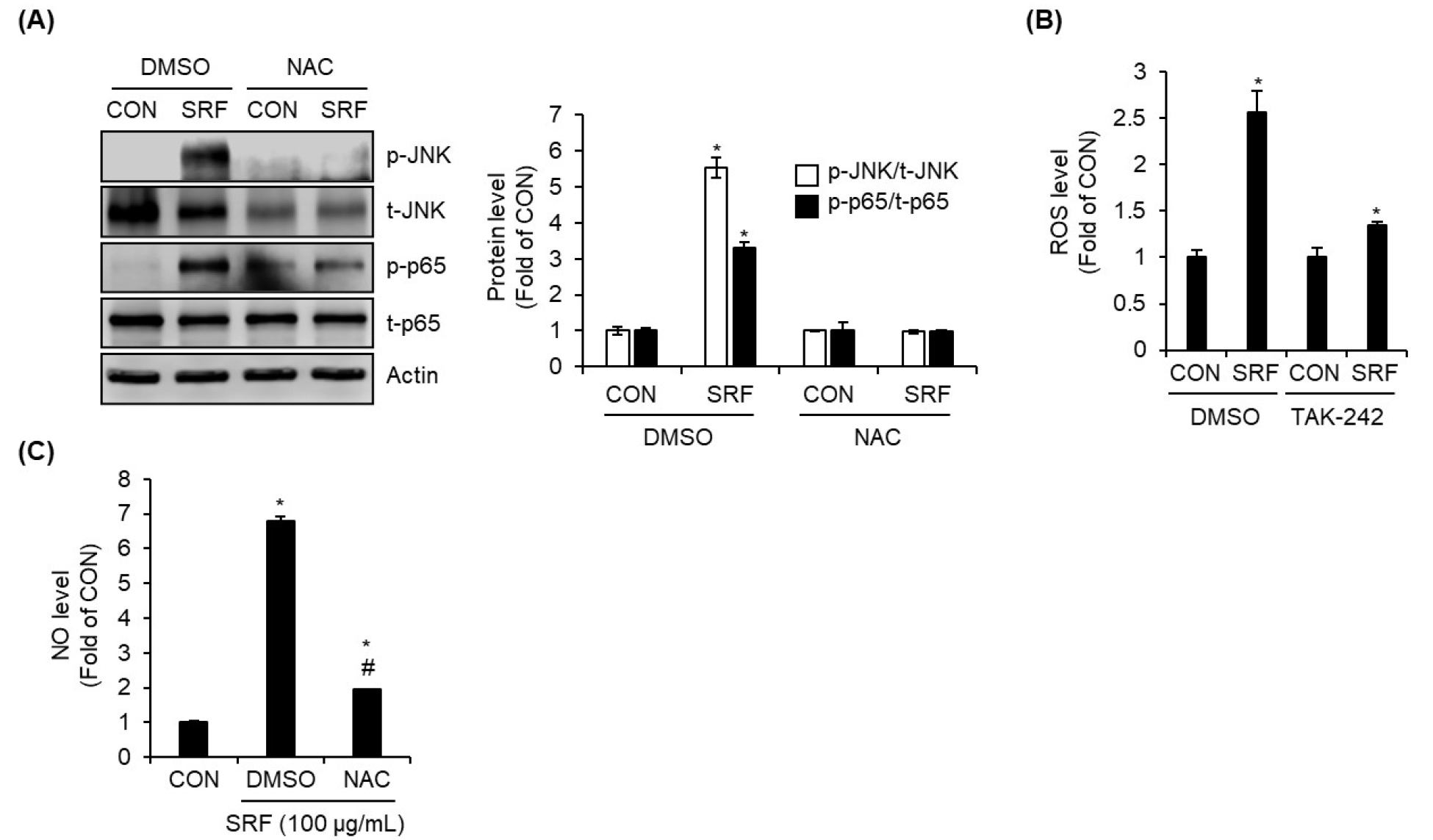
Fig. 4.
ROS are required for SRF-induced activation of JNK and NF-κB signaling and NO production in RAW264.7 cells. (A) RAW264.7 cells were pretreated with the ROS inhibitor NAC (10 mM) for 2 h and then co-treated with SRF (100 ㎍/mL) for 1 h, followed by assessment of p-JNK and p-p65 via Western blot analysis. (B) RAW264.7 cells were pretreated with TLR4 inhibitor TAK-242 (10 μM) for 2 h and then co-treated with SRF (100 ㎍/mL) for 24 h. Intracellular ROS levels were measured using an ROS Assay Kit. (C) RAW264.7 cells were pretreated with ROS inhibitor NAC (10 mM) for 2 h and then co-treated with SRF (100 ㎍/mL) for 24 h. NO level was measured by the Griess assay. *P<0.05 vs. CON (untreated group), #P<0.05 vs DMSO (SRF-treated group).
Quantification of major immunostimulatory phytochemical constituents in SRF
To identify the key constituents potentially responsible for the immunostimulatory activity of SRF, we quantified its total contents of polysaccharides, polyphenols, and flavonoids-phytochemical groups known to influence immune responses. The total polysaccharide content of SRF was determined to be 49.28 ± 9.09 ㎎ ME/g extract (Table 1). In addition, the total polyphenol and flavonoid contents were 108.86 ± 1.21 ㎎ TAE/g extract and 18.44 ± 0.08 ㎎ RE/g extract, respectively (Table 1).
Discussion
The present study demonstrated that treatment with the fruit extract of Sambucus racemosa subsp. pendula (SRF) significantly enhanced macrophage activation in a dose-dependent manner, as evidenced by increased production of NO, elevated expression of NOS, and the secretion of key pro- inflammatory cytokines including IL-1β, IL-6, and TNF-α. These observations suggest that SRF promotes a classical M1-like macrophage activation phenotype, commonly associated with heightened innate immune responses (Ghamangiz et al., 2025). NO is a hallmark effector molecule produced by activated macrophages, and its production is tightly regulated at the transcriptional level through the induction of iNOS (Xue et al., 2018). In this study, SRF markedly upregulated iNOS mRNA expression, suggesting that the observed increase in NO production is transcriptionally regulated. The coordinated increase in pro-inflammatory cytokines further supports the conclusion that SRF can activate macrophage- mediated immune responses. These cytokines play central roles in immune cell recruitment, activation, and inflammation, and are critical mediators in the host defense against pathogens (Bhol et al., 2024). In addition to cytokine secretion, SRF enhanced the phagocytic activity of RAW264.7 macrophages, a key functional parameter of innate immunity. The ability of macrophages to engulf and clear pathogens is essential for maintaining host defense (Hirayama et al., 2017), and the enhancement of phagocytosis suggests that SRF not only stimulates the production of immunoregulatory molecules but also improves effector function. Importantly, SRF did not exhibit cytotoxic effects on macrophages at the tested concentrations, indicating that its immunostimulatory effects are not secondary to cellular stress or damage.
To elucidate the upstream receptor responsible for SRF- mediated macrophage activation, we examined the roles of two major pattern recognition receptors, Toll-like receptor 2 (TLR2) and Toll-like receptor 4 (TLR4), using inhibitors such as C29 (TLR2 inhibitor) and TAK-242 (TLR4 inhibitor). Our results demonstrate that TLR4, but not TLR2, is essential for the immunostimulatory activity of SRF. Pretreatment of RAW264.7 macrophages with the TLR4-specific inhibitor TAK-242 completely abolished SRF-induced NO production, iNOS expression, and phagocytic activity, whereas the TLR2 inhibitor C29 had no such effect. These findings strongly indicate that TLR4 is the primary receptor mediating SRF-induced macrophage activation. TLR4 plays a central role in the recognition of microbial components such as lipopolysaccharide, as well as a variety of endogenous and exogenous ligands, including plant-derived polysaccharides and polyphenols (Kim et al., 2023; Yin et al., 2019). Several immunostimulatory natural products have been reported to act through TLR4, either by directly stimulating the receptor (Park et al., 2024; Wang et al., 2025; Yin et al., 2019). Our results are in line with these studies and suggest that certain bioactive constituents in SRF can activate macrophages via TLR4 engagement. The finding that TLR2 is dispensable for SRF activity further highlights the TLR4 specificity of SRF-mediated immunostimulation. TLR2 is commonly activated by bacterial lipopeptides and other microbial ligands, and while some plant extracts can co-activate TLR2 and TLR4 (Jeon et al., 2022; Yu et al., 2022), SRF appears to engage TLR4 selectively. This receptor specificity could provide advantages for targeted immunomodulation, as TLR4 activation has been associated with robust adjuvant effects in innate and adaptive immunity (Reed et al., 2016).
To further clarify the intracellular mechanisms by which SRF activates macrophages, we investigated the involvement of specific signaling pathways downstream of TLR4. Our results demonstrate that SRF-induced production of NO and expression of iNOS are critically dependent on the activation of JNK and NF-κB signaling pathways, but not on ERK1/2 or p38. The inhibition of JNK using SP600125 or NF-κB using BAY 11-7082 markedly attenuated SRF-induced NO production and iNOS expression, while inhibition of ERK1/2 or p38 had no significant effect. These results indicate that the JNK and NF-κB pathways are essential for mediating SRF- driven macrophage activation. MAPK and NF-κB are central signaling molecules involved in macrophage activation responding to immunostimulatory agents (Son et al., 2022). The upregulation of iNOS and subsequent NO production are tightly regulated by these transcriptional regulators, and their activation is commonly associated with TLR4 stimulation (Deng et al., 2015). Our phosphorylation studies confirmed that both JNK and the NF-κB p65 subunit were activated in response to SRF treatment, with phosphorylation observed as early as 1 hour post-stimulation. These temporal dynamics are consistent with a rapid TLR4-mediated immune activation mechanism. Moreover, the TLR4 inhibitor TAK-242 significantly suppressed SRF-induced phosphorylation of both p-JNK and p-p65, confirming that activation of JNK and NF-κB by SRF is dependent on upstream TLR4 signaling. These results further support our earlier findings that TLR4 functions as the primary pattern recognition receptor mediating SRF-induced macrophage activation. Taken together, these data delineate a mechanistic pathway in which SRF stimulates macrophages through a TLR4-JNK/NF-κB axis. It is noteworthy that ERK1/2 and p38 MAPK pathways often implicated in macrophage activation, were not involved in the SRF-induced production of NO. This selective activation profile suggests that SRF may preferentially engage distinct signaling branches downstream of TLR4. A possible explanation lies in the specific structure or receptor-binding affinity of SRF components, such as certain polysaccharides or polyphenols
Our findings demonstrate that ROS are essential intermediates in the SRF-TLR4 signaling cascade leading to macrophage activation. Pretreatment with the ROS scavenger, NAC, effectively suppressed SRF-induced phosphorylation of both JNK and the NF-κB p65 subunit, indicating that ROS production is required for activation of these signaling pathways. In the absence of NAC, SRF markedly enhanced the phosphorylation of both proteins, further confirming that ROS serve as a positive regulator of SRF-mediated signaling events. We further showed that SRF directly promotes intracellular ROS generation in RAW264.7 macrophages. This effect was significantly attenuated by TLR4 inhibition with TAK-242, suggesting that ROS production occurs downstream of TLR4 activation. These results are consistent with prior studies reporting that TLR4 engagement by natural polysaccharides or polyphenols can stimulate ROS generation, which subsequently modulates redox-sensitive signaling pathways such as JNK and NF-κB (Geum et al., 2020; Wang et al., 2024b). The link between TLR4 activation and ROS production may involve NADPH oxidase, mitochondria, or other oxidoreductase systems, although the precise source of ROS in SRF-treated macrophages remains to be determined (Park et al., 2004; West et al., 2011). Importantly, inhibition of ROS production by NAC led to a substantial reduction in NO production in SRF-stimulated cells, underscoring the functional significance of ROS in mediating downstream effector responses. These data position ROS as a central molecular bridge between TLR4 activation and the induction of immunostimulatory factors, aligning with the established role of ROS as secondary messengers in innate immune signaling (Lindermayr and Yildirim, 2025). The identification of ROS as a critical component of the SRF- TLR4-JNK/NF-κB axis provides mechanistic insight into how SRF orchestrates macrophage activation.
Phytochemical analysis of SRF revealed that SRF contained 49.28 ± 9.09 ㎎ ME/g extract of total polysaccharides, 108.86 ± 1.21 ㎎ TAE/g extract of total polyphenols, and 18.44 ± 0.08 ㎎ RE/g extract of total flavonoids, all of which are well-documented for their immunostimulatory properties (Cui et al., 2020; Guo et al., 2021; Lee et al., 2014; Son et al., 2022; Tabarsa et al., 2022; Wang et al., 2024b; Wang et al., 2025; Wu et al., 2025; Xu et al., 2024; Yang et al., 2022). These results suggest that multiple bioactive compound classes may collectively contribute to the immunostimulatory effects observed in RAW264.7 macrophages. Polysaccharides, particularly from medicinal plants and fruits, have been widely reported to activate macrophages via pattern recognition receptors such as TLR4, leading to enhanced cytokine production and phagocytosis (Wang et al., 2025; Wu et al., 2023). Given that SRF-mediated macrophage activation was shown to be TLR4-dependent in this study, the SRF’s polysaccharides may represent a major driver of this activity. Similarly, plant-derived polyphenols and flavonoids are known to activate immune function (Guo et al., 2021; Lee et al., 2014; Wu et al., 2025; Xu et al., 2024). These compounds can act synergistically with polysaccharides to potentiate innate immune responses.
In conclusion, this study demonstrates that SRF enhances NO production, cytokine secretion, and phagocytosis in RAW264.7 macrophages via the TLR4-ROS-JNK/NF-κB pathway. SRF contains polysaccharides, polyphenols, and flavonoids, which may contribute to its immunostimulatory activity. These findings suggest the potential of SRF as a natural immunostimulatory agent. However, this study has several limitations that should be acknowledged. First, all experiments were conducted using an in vitro macrophage model (RAW264.7 cells), which, while widely used for studying innate immune mechanisms, cannot fully recapitulate the complexity of immune responses in a living organism. Therefore, in vivo studies will be essential to validate the immunostimulatory efficacy and safety of SRF under physiological conditions. Second, although we quantified the total contents of polysaccharides, polyphenols, and flavonoids, we did not identify the specific active constituents directly responsible for SRF-induced macrophage activation. Given that the biological activity of these compound classes can vary substantially depending on their molecular structures, future studies should employ bioassay-guided fractionation and advanced analytical techniques (e.g., LC-MS/MS, NMR) to isolate and characterize the precise bioactive molecules. Such analyses will be critical for elucidating the structure-activity relationships underlying SRF’s immunostimulatory effects.
