

Introduction
Materials and Methods
Reagents and antibodies
Preparation of AH Extract
Cell culture and cytotoxicity assay
In vitro wound-healing assay
Measurement of transepithelial electrical resistance (TER)
Matrigel invasion assay
RNA extraction and reverse transcription-PCR
Protein extraction and western blot analysis
Gelatin zymographic analysis
Statistical analysis
Results
AH inhibits cell proliferation and migration of Hep3B cells
AH inhibits the cell invasion of Hep3B cells
AH suppresses MMPs activity and expression in Hep3B cells
AH increased TER values and modulated the expression levels of TJ-related genes in Hep3B cells
AH-induced anti-invasiveness by inhibition of the PI3K/AKT signaling pathway
Discussion
Introduction
Metastasis is the process by which a cancer spreads from the location at which it first arises as a primary tumor to distant locations in the body. This process involves several steps: invasion of adjacent tissues, intravasation, transport of cancer cells through the circulatory system, arrest at a secondary site, and extravasation and growth in a secondary organ. In epithelial cells, several specialized and distinct intercellular structures, including gap junctions, tight junctions (TJs), adherens junctions (AJs), and desmosomes, are responsible for establishing contact between neighboring cells. Among these, TJs, situated at the membrane between the apical and lateral regions of polarized epithelial cells selectively regulate the passage of molecules and ions via the paracellular pathway and restrict the lateral movement of molecules in the cell membrane (Schneeberger and Lynch, 2004; Soler et al., 1999). In precancerous lesions of the epithelia and cancerous epithelia, TJ strands become disorganized or lost altogether, and TJs become “leaky,” as indicated by decreased resistance to electrical current (measured by transepithelial electrical resistance; TER) and increased expression levels of markers of paracellular permeability (Furuse et al., 2001; Rubenwolf and Southgate, 2011; Soler et al., 1999). Claudins, which are major integral membrane proteins that form the backbone of TJs, can form homodimers or heterodimers to produce paired strands between adjacent cells and act as a barrier to the paracellular flux of water, solution, and the transmigration of other cells, thereby determining the characteristic permeability properties of different epithelial tissues (Angelow and Yu, 2007; Morin, 2005; Uthch et al., 2006). Recent studies have provided evidence that claudins are aberrantly expressed in various cancers and are associated with cancer development and progression, suggesting that they have key cellular functions distinct from their roles in TJ complexes (Krause et al., 2008; Singh et al., 2010; Turksen and Troy, 2011).
The matrix metalloproteinases (MMPs) form a family of zinc-dependent endopeptidases that play important roles in inflammatory tissue destruction, angiogenesis, and cancer cell metastasis. In cancer cells, these enzymes may contribute to cell invasion, favoring modifications to the matrix and resulting in tumor cell invasion (John and Tuszynski, 2001; Vihinen et al., 2005). Invasive cancer cells use MMPs to degrade the extracellular matrix (ECM) and basement membrane during metastasis. Among the human MMPs reported to date, gelatinases A (MMP-2) and B (MMP-9), which are abundantly expressed in various malignant tumors, contribute to cancer invasion and metastasis. Generally, MMP-2 (72 kDa) is preferentially secreted by fibroblasts and various epithelial cells, whereas MMP-9 (92 kDa) is preferentially expressed by inflammatory cells; both are frequently associated with the invasive metastatic potential of tumor cells (Gibbs et al., 1999; Mook et al., 2004). Tissue inhibitors of metalloproteinases (TIMPs), which are naturally occurring inhibitors of MMPs, play an important role in the complex regulation of MMPs. They inhibit the catalytic activity of MMPs by binding to activated MMPs and controlling ECM breakdown (Lambert et al., 2004; Uzui et al., 2002). Thus, the balance between MMPs and TIMPs plays a vital role in maintaining the integrity of healthy tissues. MMP inhibitors and TIMP activators are expected to be useful chemotherapeutic agents for the treatment of malignancies.
Members of the Alnus species have been used in several traditional medicines, such as cathartics, emetics, galactogogues, febrifuges, hemostatics, parasiticides, vermifuges, skin tonics, and astringents (Guo et al., 2001a). Alnus hirsuta (Turcz. ex Spach) Rupr. (AH) is geographically distributed in Korea, Japan, Northeast China, and Russia. Its bark has been used as an antipyretic, expectorant, anti-asthmatic, and tea-based treatment for alcoholism (Lee, 1996). Previous studies on the chemical constituents of Alnus species have led to the isolation of various tannins, flavonoids, diarylheptanoids, and triterpenoids (Aoki et al., 1990; Choi et al., 2012; Jeong et al., 2000; Lee et al., 1992, 1999; Suga et al., 1972; Terazawa et al., 1984). These studies have shown that members of Alnus species are a good source of diarylheptanoids and that plants of this genus exhibit anti-oxidative, anti-inflammatory, anti-atopic, anti-bacterial, and anti-adipogenic activities (Choi et al., 2012; Joo et al., 2009; Lee et al., 2010, 2013). However, the molecular mechanisms underlying the anti-metastatic effects of AH are not completely understood.
Therefore, in this study, using Hep3B hepatocellular carcinoma cells, we investigated the anti-metastatic potential of AH and the underlying intracellular signal transduction pathways involved in metastasis inhibition. The results of this study demonstrated that AH inhibits two aspects of metastatic potential, namely, cell motility and invasiveness, by modulating the levels of TJ-associated factors and the activities of MMPs.
Materials and Methods
Reagents and antibodies
Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), and the CyQUANTTM LDH cytotoxicity assay kit (C20300) were purchased from Thermo Fisher Scientific, Inc. (Waltham, MA, USA). Antibiotics were purchased from Welgene (Daegu, Korea). 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium (MTT), Matrigel, eosin (H&E), and lipopolysaccharide (LPS) were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). Antibodies against claudin-1 (519000), -2 (516100), -3 (341700), and -4 (329400) were purchased from Calbiochem (San Diego, CA, USA). Akt (sc-8312), phosphorylated Akt (pAkt) (sc-101629), tissue inhibitors of metalloproteinase (TIMP)-1 (sc-5538) and TIMP-2 (sc-5539), MMP-2 (sc-10736), MMP-9 (sc-10737), and actin (sc-70319) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Peroxidase-labeled donkey anti-rabbit and sheep anti-mouse immunoglobulins were purchased from Amersham Corp. (Arlington Heights, IL, USA). All other chemicals were purchased from Sigma-Aldrich Chemical Co.
Preparation of AH Extract
To prepare an aqueous extract of AH, the bark of AH was washed thoroughly in running water, air-dried at room temperature, and ground to powder using a mechanical grinder. After adding 2 L of distilled water per 100 g of bark part of AH, the mixture was boiled at 110℃ for 2 h in a heater equipped with a reflux-cooling device. After centrifuging at 3,000 rpm for 20 min, the residue was removed, the supernatant was filtered using a Whatman filter (No. 2), and a solid extract was produced through a decompression evaporation process. The solid component was then pulverized using a mortar and pestle and stored at -70℃. A stock solution of AH (100 ㎎/mL) was prepared using dimethyl sulfoxide (DMSO) as the solvent and was used to prepare dilutions for experimentation.
Cell culture and cytotoxicity assay
The human HCC cell line Hep3B (p53 null type; ATCC; American Type Culture Collection) was cultured in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum, 100 U/mL penicillin, and 100 µg/mL streptomycin. The cells were cultured in a humidified chamber at 37℃ with 5% CO2. Cytotoxicity was assessed using the standard MTT and lactate dehydrogenase (LDH) assays. Briefly, Hep3B cells were seeded in 6-well plates at a density of 3 × 105 cells/well. After 24 h, the cells were treated with various concentrations of AH (0-10 ㎍/mL) for 24 h. The cells were then incubated with MTT solution (0.5 ㎎/mL) for 3 h at 37℃ in the dark, the medium was removed, and the formazan precipitate was dissolved in DMSO. Absorbance of the formazan product was measured at 540 ㎚ using a Cytation-3 microplate reader (BioTek, Shoreline, WA, USA).
For the LDH cytotoxicity assay, sample treatment was performed identically to the MTT assay described previously, and an LDH cytotoxicity assay kit (Thermo Fisher Scientific, Waltham, MA, USA) was used according to the manufacturer’s instructions. At the end of 24 h of incubation, 50 μL of supernatant was transferred from each well to a new plate, and a 50-μL reaction mixture was added. The plates were then incubated in the dark at room temperature for 30 min. Then, 50 μL of stop solution was added to each well, and measurements were recorded at wavelengths of 490 and 680 ㎚ with a microplate reader.
In vitro wound-healing assay
Hep3B cells were grown to confluence on 30-mm cell culture dishes coated with 20 µg/mL of rat tail collagen (BD Biosciences, Bedford, MA, USA). The confluent cells were wounded by scraping with a pipette tip. After wounding, the cultures were washed twice with PBS, and the control cells were exposed to medium alone. The cells were incubated in 1% FBS-containing medium supplemented with various concentrations of AH for 24 h. Cell wound closure was observed and photographed under a microscope at ×40 magnification. The culture treatments were repeated twice, and each sample was assayed in triplicate (Zhu et al., 2011).
Measurement of transepithelial electrical resistance (TER)
Transepithelial electrical resistance (TER, a measure of tight junction formation) was measured using an Epithelial Tissue Voltohmmeter (EVOM; World Precision Instruments, FL, USA) equipped with a pair of STX-2 chopstick electrodes. Briefly, Hep3B cells were seeded into the 8.0 μm pore size insert (upper chamber) of a Transwell® (Corning Costar Corp., NY, USA) and allowed to reach full confluence, following which fresh medium was replaced for subsequent experiments. Inserts without cells, inserts with cells in the medium, and inserts with cells with AH were treated for 24 h. Electrodes were placed in the upper and lower chambers, and resistance was measured with the EVOM (Grant-Tschudy and Wira, 2005).
Matrigel invasion assay
Hep3B cells were incubated in DMEM supplemented with 10% FBS and collected by trypsinization. Cells (2 × 105 cells/well) in serum-free medium were added to the inner cup of a 24-well Transwell chamber (Corning Life Sciences, Oneonta, NY, USA) that had been coated with 50 mL of Matrigel (BD Biosciences, Franklin Lakes, NJ, USA; 1:10 dilution in serum-free medium). The medium supplemented with 10% serum or the indicated agent was added to the outer cup. After 24 h, cells that had migrated through the Matrigel and the 8-mm pore size membrane were fixed, stained with hematoxylin and eosin (H&E, Sigma-Aldrich Chemical Co.), and photographed under an inverted microscope. Each experiment was performed in triplicate.
RNA extraction and reverse transcription-PCR
Total RNA was prepared using RNeasy kit (Qiagen, La Jolla, CA, USA) and primed with random hexamers for synthesis of complementary DNA using AMV reverse transcriptase (Amersham Corp., Arlington Heights, IL, USA), according to the manufacturer's instructions, using DNAse I (1 U/㎍ RNA) pretreated total mRNA. Polymerase chain reaction (PCR) was performed in a Mastercycler (Eppendorf, Hamburg, Germany) using the primers listed in Table 1. Conditions for PCR reactions were 1× (94℃ for 3 min), 35× (94℃ for 45 s, 58℃ for 45 s, and 72℃ for 1 min), and 1× (72℃ for 10 min). The PCR amplification products were electrophoretically separated on a 1% agarose gel and visualized via ethidium bromide (EtBr) staining.
Table 1.
Sequence of primers used for RT-PCR
Protein extraction and western blot analysis
Total cell lysates were prepared in an extraction buffer comprising the following: 25 mM Tris-Cl (pH 7.5), 250 mM NaCl, 5 mM ethylendiaminetetra acetic acid, 1% nonidet P-40, 0.1 mM sodium orthovanadate, 2 µg/mL leupeptin, and 100 µg/mL phenylmethylsulfonyl fluoride. Protein concentrations were determined using a Bio-Rad protein assay kit (Bio-Rad Laboratories, Hercules, CA, USA). For western blot analysis, proteins (30-50 µg) were separated using 8-10% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis and then electrotransferred to a nitrocellulose membrane (Schleicher & Schuell, Keene, NH, USA). Membranes were blocked with 5% skim milk for 1 h and subjected to immunoblotting using the desired antibodies. Proteins were visualized using the enhanced chemiluminescence (ECL) method, according to the recommended procedure (Amersham Co.). Primary antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA) and Calbiochem (Cambridge, MA, USA). Peroxidase-labeled donkey anti-rabbit and sheep anti-mouse immunoglobulins were obtained (Amersham Co.).
Gelatin zymographic analysis
Following incubation with AH for 24 h, the cell culture supernatants were collected and centrifuged at 400 × g for 5 min. The cell-free supernatant was mixed with 2X sample buffer (Invitrogen), and zymography was performed using precast gels (10% polyacrylamide and 0.1% gelatin). Following electrophoresis, gels were washed twice at room temperature for 30 min in 2.5% Triton X-100; washed in buffer containing 50 mM Tris-HCl, 150 mM NaCl, 5 mM CaCl2, 1 µM ZnCl2, and 0.02% NaN3 at pH 7.5; and incubated in this buffer at 37℃ for 24 h. Thereafter, gels were stained with 0.5% (w/v) Coomassie brilliant blue G-250 (Bio-Rad) for 1 h and then lightly destained in methanol/acetic acid/water (3:1:6). Clear bands appeared on the Coomassie-stained blue background in areas of gelatinolytic activity. Gels were scanned, and images were processed by extracting the blue channel signal, convertting it to black and white, and inverting it for the quantification of gelatinolytic activities from the integrated optical density (Song et al., 2011).
Statistical analysis
Data are shown as the mean ± SEM and were analyzed using one-way analysis of variance (ANOVA) followed by Tukey's HSD test using Prism ver. 9.2 (GraphPad, CA, USA). The level of significance was set at p < 0.05.
Results
AH inhibits cell proliferation and migration of Hep3B cells
To investigate the effect of AH on the viability of Hep3B cells, the cells were treated with various concentrations of AH for 24 h and subjected to MTT and LDH assays. When compared with the control, treatment with 8 µg/mL AH caused approximately 79% inhibition of cell growth, and the same cytotoxicity assessment based on the amount of lactate dehydrogenase (LDH) released into the medium revealed that treatment with 8 µg/mL AH was cytotoxic. However, AH in the range of 2-6 µg/mL did not have a significant cytotoxic effect on Hep3B cells (Fig. 1A and B). Therefore, AH concentrations within this lower range were used in subsequent experiments. To investigate the inhibitory effect of AH on the migration of Hep3B cells, an in vitro wound-healing assay was performed. Results demonstrated that 2-6 µg/mL AH, which was not cytotoxic, as shown by MTT assay dose-dependently, delayed the motility of Hep3B cells compared to that of control cells (Fig. 1C and D).
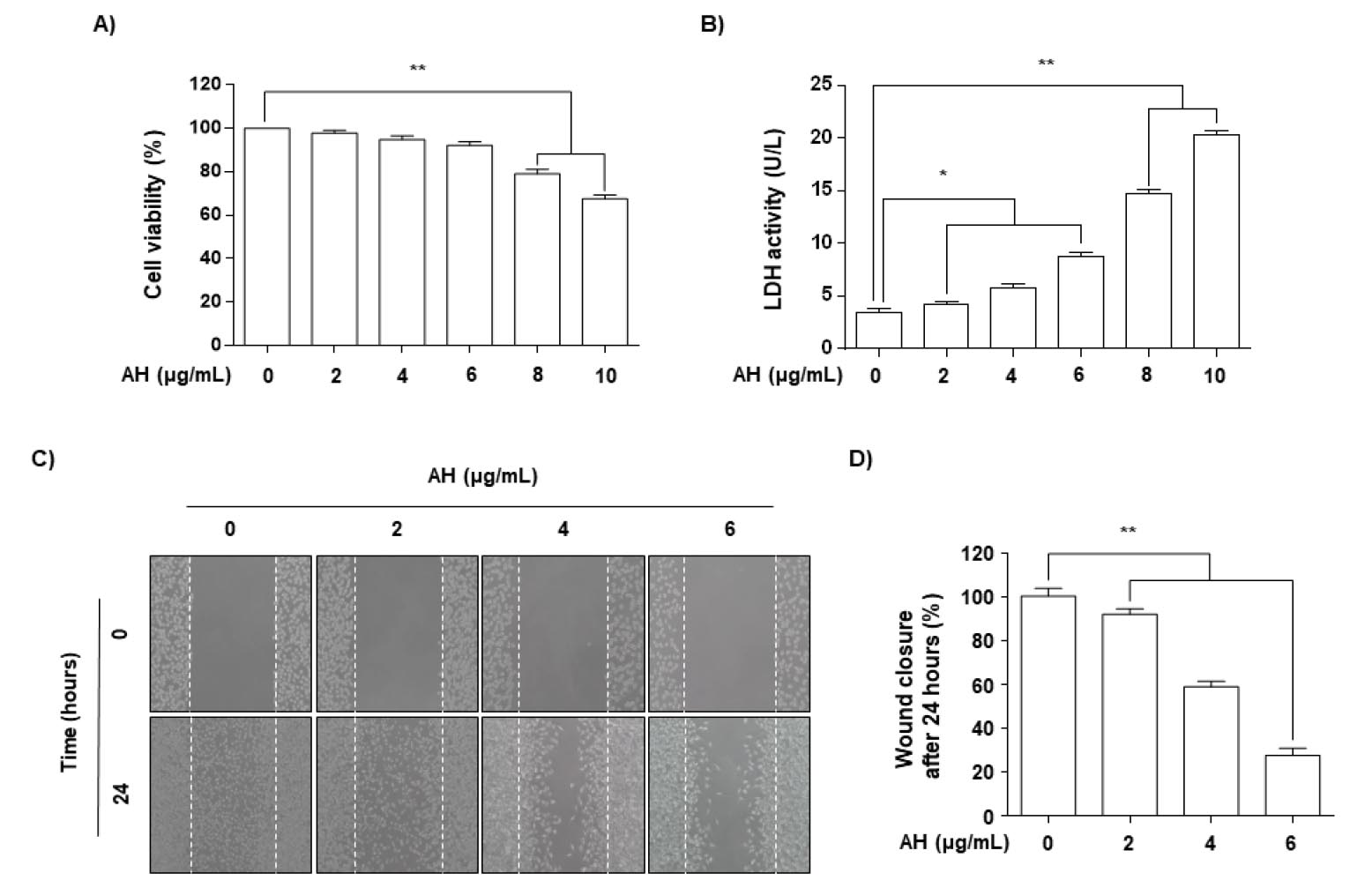
Fig. 1.
Effects of AH on cell viability and motility in Hep3B cells. (A and B) Cells were seeded at an initial density of 3×105 cells/well, incubated for 24 h, and treated with various concentrations of AH for 24 h. Cell viability was measured using an MTT and LDH assay. Each point represents the mean ± SD of three independent experiments. A Student's t-test was used for determination of significance (**p<0.01 vs. untreated control). (C and D) Cells were kept on 6-well plates for 24 h before a wound was created by a yellow pipette tip to scrape the confluent cell layers. AH at various concentrations was added to the wells, and cells were then incubated for 24 h. (C) Some of the representative photographs of invading treated and untreated cells are presented. (D) Percentage of inhibition of cell migration was quantified by manual counting, and six randomly chosen fields were analyzed for each well. Each point represents the mean ± SD of three independent experiments (**p<0.01 vs. control group).
AH inhibits the cell invasion of Hep3B cells
Using a Boyden chamber invasion assay, we attempted to determine whether the inhibitory effects of AH were associated with decreased cell invasion. As shown in Fig. 2, AH treatment markedly reduced cell invasion through the Matrigel chamber in a concentration-dependent manner, suggesting that the inhibitory effects on cell migration were associated with the inhibition of invasive activity in LNCaP cells.
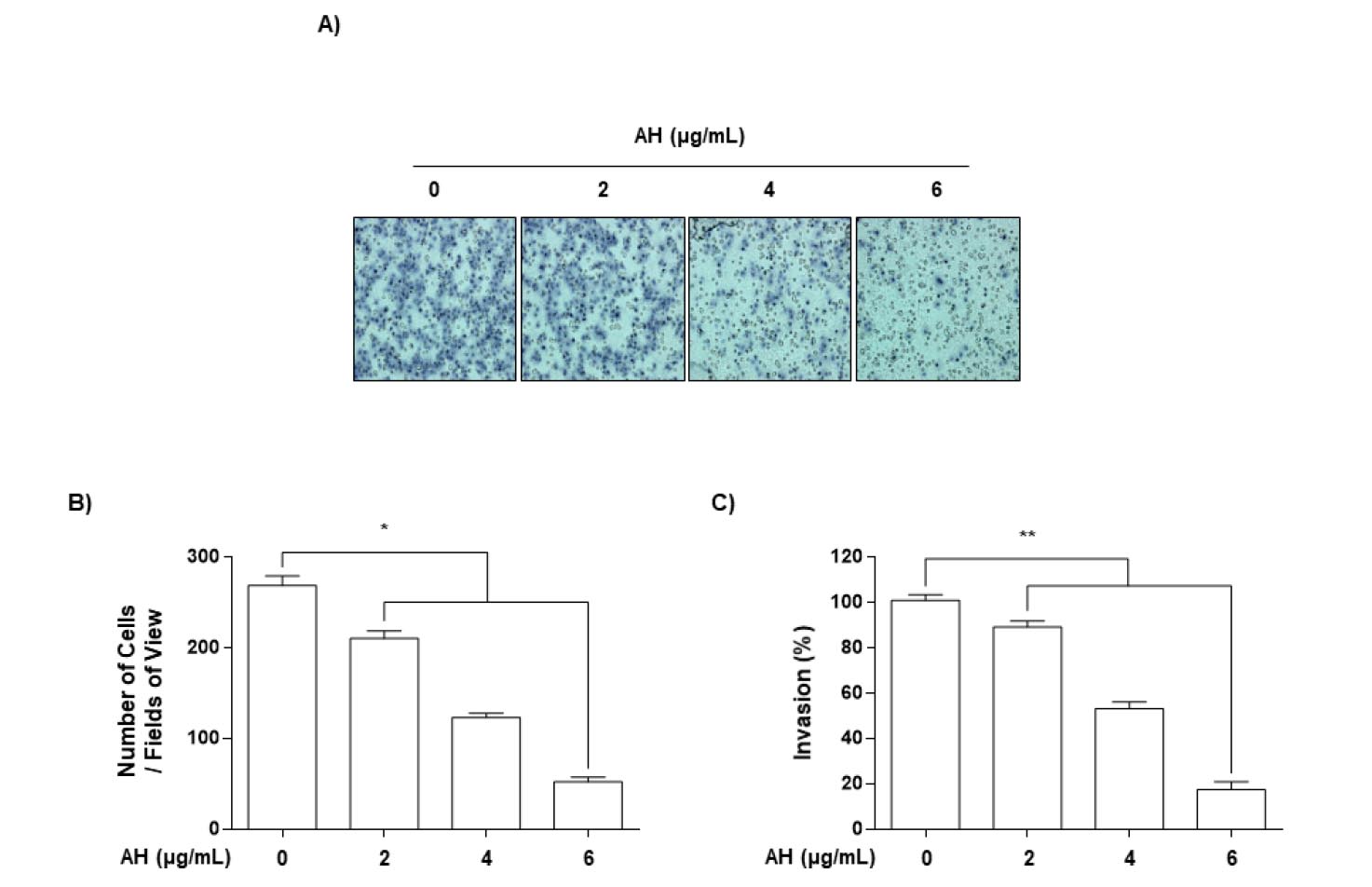
Fig. 2.
AH inhibits invasion of Hep3B cells. Cells pretreated with the indicated concentrations of AH for 24 h were plated onto the apical side of Matrigel-coated filters in serum-free medium containing either vehicle or AH. Medium containing 3% FBS was placed in the basolateral chamber to act as a chemoattractant. After 24 h, cells on the apical side were wiped off using a Q-tip. Next, (A) cells on the bottom of the filter were stained using H&E, and then (B) counted. (C) Rates of invasion were measured at 560 nm wavelength by an ELISA reader. Results are in mean ± SD values obtained from three independent experiments (*p<0.05 and **p<0.01 vs. control group).
AH suppresses MMPs activity and expression in Hep3B cells
Because Matrix metalloproteinases (MMPs) activation is crucial for extracellular matrix (ECM) degradation, which is required for metastasis (John and Tuszynski, 2001; Vihinen et al., 2005), we used gelatin zymography, RT-PCR, and western blot analyses to assess whether AH regulates the activation and expression of MMP-2 and -9 in Hep3B cells. As indicated in Fig. 3A and B, AH inhibited the activities of MMP-2 and -9 and that the effects occurred in a dose-dependent manner (Fig. 3C and D). In contrast, AH treatment increased the levels of TIMP-1 and -2 mRNA and protein in a concentration-dependent manner compared to the control group (Fig. 3C and D). These results indicate that the anti-invasive effect of AH is associated with increased TIMP levels, as well as the inhibition of both enzyme activity and expression of MMP-2 and -9 in Hep3B cells.
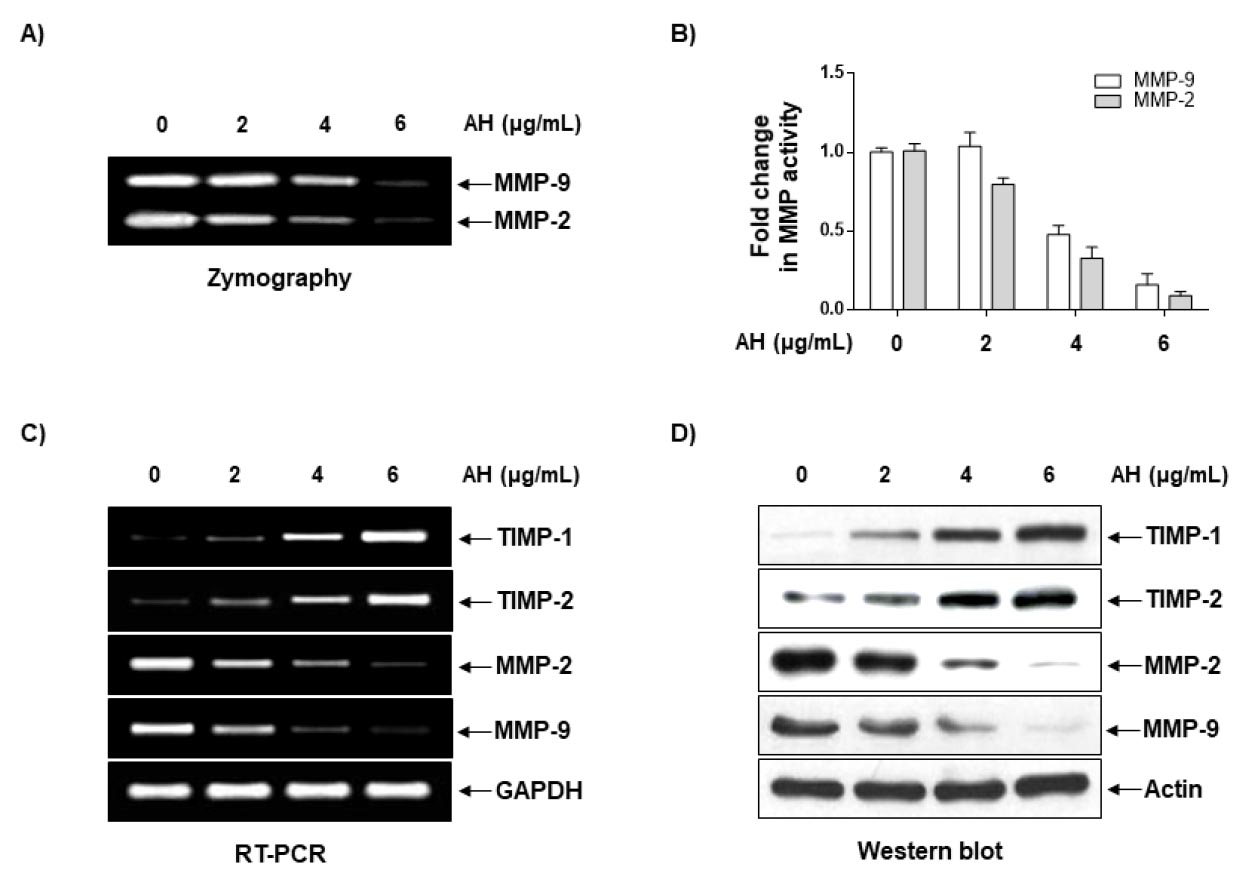
Fig. 3.
AH suppresses activities and levels of MMP-2 and -9 and increase levels of TIMP-1 and -2 in Hep3B cells. (A) Cells were treated with the indicated concentrations of AH for 24 h. The medium was collected, and activities of MMP-2 and -9 were measured by zymography, and then (B) counted. (C) Total RNA was isolated from cells grown under the same conditions as (A) and reverse-transcribed. Resulting cDNAs were then subjected to the polymerase chain reaction. The reaction products were run on 1% agarose gel electrophoresis and visualized by ethidium bromide staining. GAPDH was used as the internal control. (D) Cells were sampled and lysed, and 30 ㎍ of proteins were separated by electrophoresis on SDS-polyacrylamide gels. Western blotting was then performed using the indicated antibodies and an ECL detection system. β-actin was used as an internal control.
AH increased TER values and modulated the expression levels of TJ-related genes in Hep3B cells
Since altered TJs lead to a decrease in resistance to electrical current (as measured by TER) and an increase in paracellular permeability (Soler et al., 1999; Utech et al., 2006), TER values were measured to determine the interaction between the tightening and the anti-invasive activity of AH. The results shown in Fig. 4A indicate that TER values increased substantially in response to increasing concentrations of AH, suggesting that AH increases the tightening in Hep3B cells. To elucidate the mechanism by which AH enhances TJ activity and reduces invasive activity, we determined the levels of TJ claudin regulators via RT-PCR and western blotting. As shown in Fig. 4B and C, the expression of claudins (claudin-1, -2, -3, and -4) was downregulated in a dose-dependent manner in AH-treated cells, suggesting that this modulation contributed to TJ tightening.
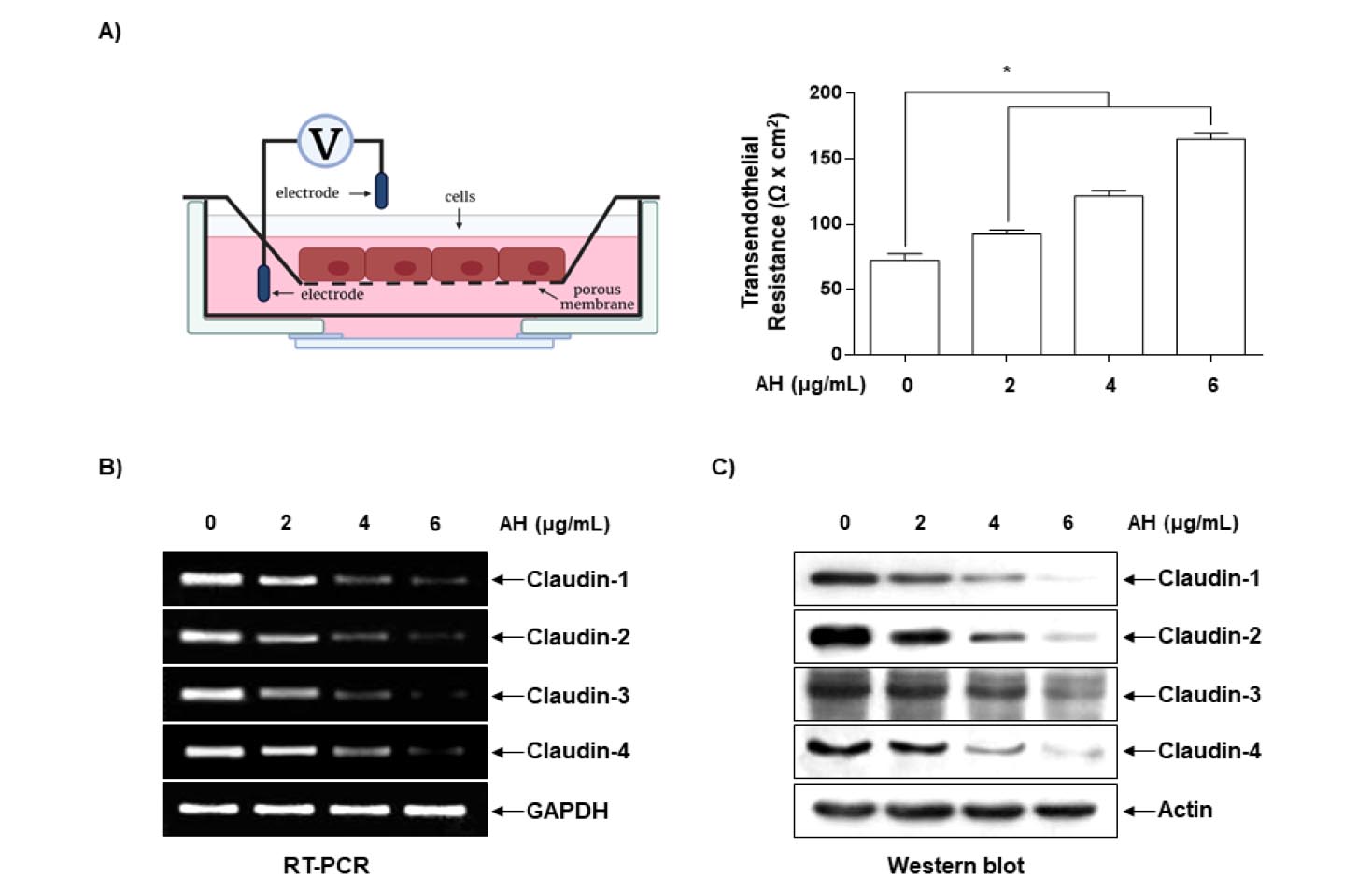
Fig. 4.
Effects of AH on TER values and expression of claudins in Hep3B cells. (A) Cells were plated onto trans-wells and grown in media, and AH was then added to both the apical and basolateral compartments in triplicate. TER values were measured using an EVOM. Each point represents the mean ± SD of three independent experiments (*p<0.05 vs. control group). (B) Total RNA was isolated from cells grown under the same conditions as (A) and reverse-transcribed. Resulting cDNAs were then subjected to the polymerase chain reaction. The reaction products were run on 1% agarose gel electrophoresis and visualized by ethidium bromide staining. GAPDH was used as the internal control. (C) Cells were sampled and lysed, and 30 ㎍ of proteins were subjected to Western blot analyses using the indicated antibodies and an ECL detection system. β-actin was used as an internal control.
AH-induced anti-invasiveness by inhibition of the PI3K/AKT signaling pathway
Because several lines of evidence have implicated the PI3K/AKT signaling pathway in the expression of MMPs and induction of cancer cell metastasis (Li et al., 2014; Park et al., 2012), we assessed the changes in the phosphorylation of AKT, a downstream target of PI3K, after AH treatment. Western blotting indicated that AH treatment significantly reduced the relative expression levels of pAKT in a concentration-dependent manner; however, the levels of total AKT protein remained unchanged (Fig. 5A).
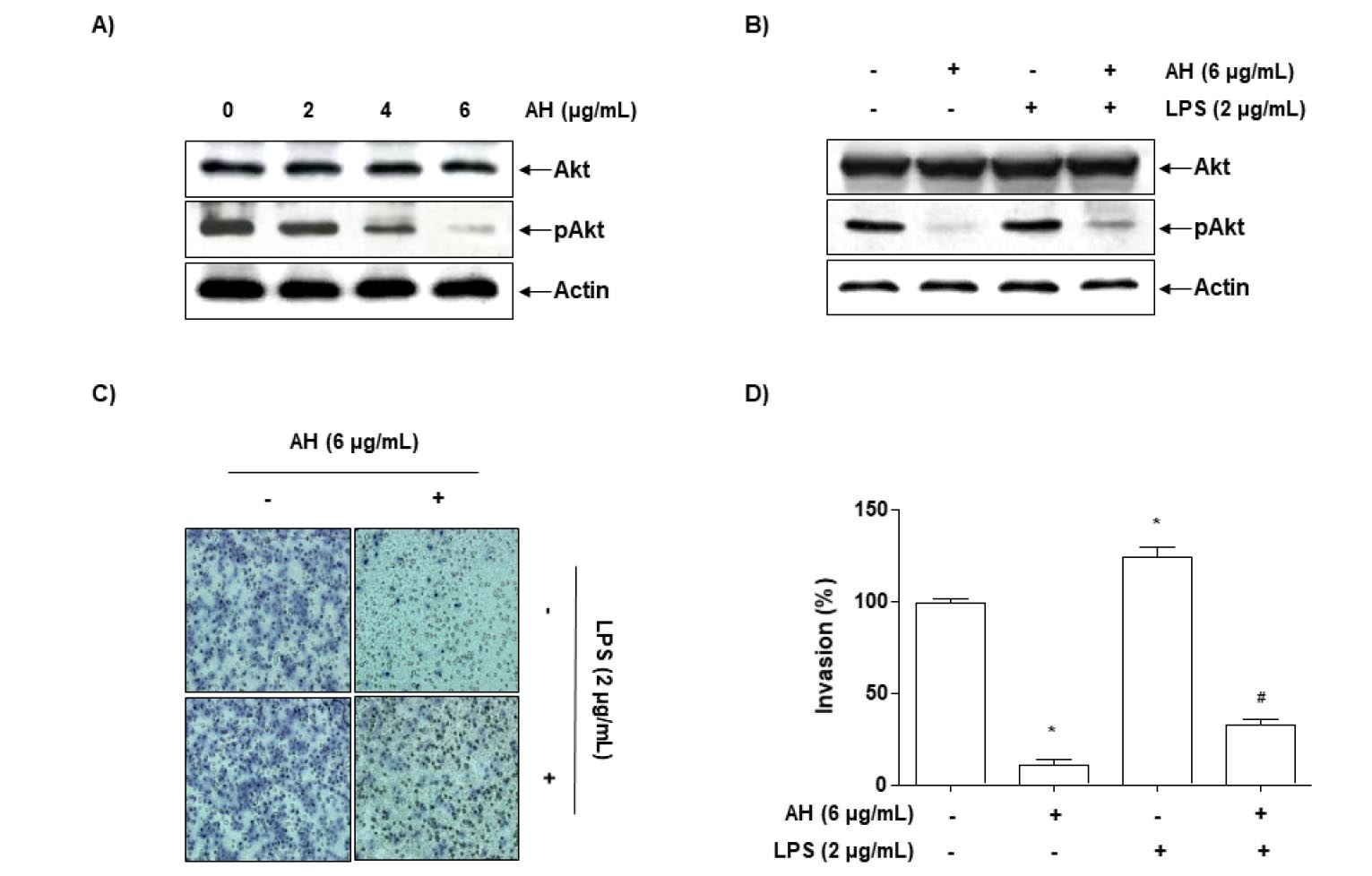
Fig. 5.
AH attenuates LPS-induced Akt phosphorylation and cell invasion in Hep3B cells. Cells were treated with the indicated concentrations of AH for 24 h (A) or incubated with 6 ㎍/mL AH for 1 h before treatment with 2 ㎍/mL LPS for 24 h (B). Cellular proteins were separated by SDS-polyacrylamide gels and transferred onto nitrocellulose membranes. Immunoblotting analyses were performed with anti-Akt and anti-p-Akt antibodies and an ECL detection system. actin was used as an internal control. (C, D) Cells treated with 6 ㎍/mL AH for 1 h before being challenged with LPS (2 ㎍/mL) for 24 h were plated onto the apical side of Matrigel-coated filters in serum-free medium containing either vehicle or AH. Medium containing 10% FBS was placed in the basolateral chamber to act as a chemoattractant. After 24 h, cells on the apical side were wiped off using a Q-tip. Next, cells on the bottom of the filter were stained using H&E, and rates of invasion were then measured at 560 nm wavelength by an ELISA reader. The results are in mean ± SD values obtained from three independent experiments (*p<0.05 vs. control group; #p<0.05 vs. AH-treated group).
LPS, a major component of the outer membrane of gram-negative bacteria, is one of the most potent activators of the PI3K/AKT signaling pathway for the stimulation of cancer cell metastasis, and its activation mechanism is relatively well established (Hsu et al., 2011; O’Leary et al., 2012). Accordingly, we evaluated whether AH regulates the LPS-induced activation of the PI3K/AKT signaling pathway. As shown in Fig. 5B, stimulation with LPS significantly increased AKT phosphorylation; however, pretreatment with AH abolished AKT phosphorylation. The results of parallel experiments showed that pretreatment with AH inhibited the LPS-induced invasion of Hep3B cells (Fig. 5C and D), indicating that AH-induced inhibition of invasion is mediated through suppression of the PI3K/Akt pathway.
Discussion
Different plant components are rich sources that are far from being well utilized, and chemical constituents from plants demonstrate various significant bioactivities. AH is a deciduous tree of the family Betulaceae and genus Alnus. More than 15 species are native to Korea and are found throughout North Korea, China, Russia, and Japan (Lee, 1966; Oh et al., 2006, 2008). In oriental medicine, the bark of AH is called saegjeog-yang and has been used to treat heat, hemorrhage, diarrhea, gastrointestinal disorders, lymphatic diseases, and cancer (Guo et al., 2001b); however, its antimetastatic activity and associated mechanisms in hepatocellular carcinoma cells remain unclear. Therefore, we investigated the mechanisms underlying this phenomenon and found that AH significantly reduced the invasive and metastatic abilities of Hep3B cells.
Metastasis, the main cause of death in cancer patients, is a complex multistep process involving cell adhesion, invasion, and migration. MMPs, which are highly expressed in various malignant tumors, play an important role in cell motility and invasion via the degradation of ECM components in blood or lymph vessels (Hanahan and Weinberg, 2011; Khasigov et al., 2003). Gelatinase MMPs, such as MMP-2 and -9, promote tumor cell invasion in various cancer cell lines because of their ability to degrade various types of collagen (Egeblad and Werb, 2002; Vu and Werb, 2000). MMP activity is tightly controlled by TIMPs, which form complexes with MMPs to inhibit the active forms of enzymes (Kessenbrock et al., 2010; Khasigov et al., 2003). To elucidate the mechanism underlying the antimetastatic effect of AH, we examined the activities of MMP-2 and -9 in Hep3B cells treated with AH. AH significantly inhibited the activity of both MMP-2 and -9, as determined by gelatin zymography (Fig. 3A and B). Our results also showed that AH markedly inhibited the mRNA and protein expression of MMP-2 and -9; however, the levels of both TIMP-1 and -2 exhibited concentration-dependent upregulation in response to AH treatment (Fig. 3C and D). Therefore, these results indicate that AH promotes an increase in the TIMP/MMP ratio as a key factor in the regulation of the antimetastatic process, which may subsequently block the breakdown of the ECM and lead to the inhibition of cell invasion.
However, there is an association between the loss of cell-cell adhesion structures and metastasis in many cancers. In precancerous lesions, tissue remodeling by disassembled and disorganized TJs, as determined by decreased resistance to TER, causes loss of cell polarity and, in turn, promotes cancer cell motility and invasiveness (Soler et al., 1999; Utech et al., 2006). The components of TJs have been well characterized, particularly those of the claudin family, which include transmembrane proteins and their extracellular domains. The claudin family of proteins forms the backbone of TJs, which are directly involved in the barrier and adhesive functions of cells to regulate paracellular permeability (Morin, 2005; Tsukita and Furuse, 2000). Several reports have shown that the TER in some tumor tissues is significantly lower than that in normal tissue, and that transepithelial paracellular permeability is higher than that in normal tissue, confirming the loss of TJ function (Morin, 2005; Soler et al., 1999; Tsukita and Furuse, 2000). Recent studies have shown that claudins are aberrantly overexpressed in various human cancers, including hepatocellular carcinoma (HCC), and are associated with the development and progression of cancer metastasis (An et al., 2020; Du et al., 2021; Mattiolo et al., 2023). These observations suggest that claudins may serve as useful biomarkers for detecting and diagnosing certain cancers. Therefore, we measured TER values to examine the relationship between TJ remodeling and the anti-invasive activity of AH and found that incubating Hep3B cells with AH increased TER in a concentration-dependent manner (Fig. 4A). In addition, AH treatment markedly suppressed the levels of claudins (-1, -2, -3, and -4) (Fig. 5B, 5C), indicating that the downregulation of claudin expression by AH was associated with increased TJ tightening in Hep3B cells.
By regulating the transcriptional activity of MMPs, numerous cell signaling pathways play critical roles in the regulation of cancer cell migration and invasion (Li et al., 2014; Park et al., 2012). For example, the activation of the PI3K/AKT signaling pathway is associated with the expression of MMP-2 and -9 in cancerous tissues, where cancer cells promote neovascularization for invasion and metastasis (Dilly et al., 2013; Liu et al., 2015). To this end, we investigated whether the anti-invasive effects of AH were associated with the inactivation of the PI3K/AKT signaling pathway and found that AH itself significantly reduced AKT phosphorylation (Fig. 5A). In addition, LPS stimulation significantly increased AKT phosphorylation; however, pretreatment with AH downregulated AKT phosphorylation (Fig. 5B). The Matrigel assay showed that AH significantly suppressed LPS-induced Hep3B cell invasion. These results indicate that AH inhibits the LPS-induced activation of the PI3K/AKT signaling pathway, which may lead to the suppression of Hep3B cell migration and invasion.
In summary, this study found that AH significantly inhibited the migration and invasion of hepatocellular carcinoma Hep3B cells by inhibiting and suppressing the expression and activity, respectively, of MMP-2 and -9. The data also show that AH increases TJ tightening associated with the downregulation of claudin expression, which is associated with inactivation of the PI3K/AKT signaling pathway. Thus, the findings of this study indicate that AH is a potential candidate for the development of chemotherapeutic treatments for hepatocellular carcinoma.
